

Volume 15, Issue 4 (July & August 2024)
BCN 2024, 15(4): 541-552 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Alijanpour S, Ghafouri-Fard S, Tonekaboni S H, Karimzadeh P, Ahmadabadi F, Rahimian E, et al . Recurrence of Developmental and Epileptic Encephalopathy9 (DEE9) Due to Parental Germline Mosaicism and a Literature Review. BCN 2024; 15 (4) :541-552
URL: http://bcn.iums.ac.ir/article-1-2697-en.html
URL: http://bcn.iums.ac.ir/article-1-2697-en.html
Sahar Alijanpour1 

 , Soudeh Ghafouri-Fard1 , Seyed Hassan Tonekaboni2 , Parvaneh Karimzadeh2 , Farzad Ahmadabadi2 , Elham Rahimian3 , Samareh Panjeshahi1 , Mohammad Miryounesi *1
, Soudeh Ghafouri-Fard1 , Seyed Hassan Tonekaboni2 , Parvaneh Karimzadeh2 , Farzad Ahmadabadi2 , Elham Rahimian3 , Samareh Panjeshahi1 , Mohammad Miryounesi *1
, Soudeh Ghafouri-Fard1 , Seyed Hassan Tonekaboni2 , Parvaneh Karimzadeh2 , Farzad Ahmadabadi2 , Elham Rahimian3 , Samareh Panjeshahi1 , Mohammad Miryounesi *1
1- Department of Medical Genetics, School of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
2- Department of Pediatric Neurology, School of Medicine, Mofid Children’s Hospital, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
3- Haghighat Imaging and Research Center, Tehran, Iran.
2- Department of Pediatric Neurology, School of Medicine, Mofid Children’s Hospital, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
3- Haghighat Imaging and Research Center, Tehran, Iran.
Keywords: Developmental and epileptic encephalopathy 9 (DEE9), Epilepsy, PCDH19, Genetic, Germline mosaicism
Full-Text [PDF 1450 kb]
| Abstract (HTML)
Full-Text:
1. Introduction
Epilepsy is one of the most common neurological conditions among children, and its highest incidence is seen in the first year of life (Fine & Wirrell, 2020). This is a heterogeneous group of disorders characterized by the triad of early-onset epileptic seizures, abnormal EEG activity, and developmental impairment, which are called developmental and epileptic encephalopathies (DEEs). They may be caused by both genetic and non-genetic etiologies (Guerrini et al., 2023). We have different types of DEEs, and one of them is developmental and epileptic encephalopathy 9 (DEE9), which results from pathogenic variants in the PCDH19 gene (Kolc et al., 2019). Several names have been adopted to signify the clinical characterization of this disorder, including girls clustering epilepsy (PCDH19- GCE) (Kolc et al., 2019), ‘‘early infantile epileptic encephalopathy-9” (EIE19) or developmental and epileptic encephalopathy 9 (DEE9), and PCDH19 clustering epilepsy (PCDH19-CE) (Depienne et al., 2009; Kolc et al., 2020). Here, we use DEE9 (OMIM #300088) to refer to the disorder.
Symptoms of DEE9 patients evolve in early infancy, mostly within the first year of life. It is often provoked by fever and presents clustered seizures, different degrees of cognitive impairment, and behavioral problems, such as autism spectrum disorder (ASD), hyperactivity, attention deficit, and aggression (Kolc et al., 2020). The clinical characteristics may be similar to those of Dravet syndrome (Depienne et al., 2009). The disorder is defined by a distinctive X-linked inheritance pattern where heterozygous females or rarely mosaic hemizygous males are affected, but hemizygous males and homozygous females are asymptomatic. PCDH19 is located in the region exposed to X-chromosome inactivation. Random X-inactivation and consequent tissue mosaicism are posited to be implicated in the pathogenesis of heterozygous females (Depienne et al., 2009; Depienne & Leguern, 2012). Although the exact underlying mechanism is still unclear, cellular interference has been proposed to clarify this unusual inheritance pattern (Depienne et al., 2009; Depienne & Leguern, 2012; Samanta, 2020; Thiffault et al., 2016). The cellular interference hypothesis postulates that the existence of a variety of cells in the brain, expressing either mutant or wild-type PCDH19 protein, results in abnormal neurodevelopment and is therefore the cardinal cause of the clinical presentations (Depienne et al., 2009).
The causative gene of DEE9 was mapped to the X chromosome in 1997 (Ryan et al., 1997) and determined to be PCDH19 in 2008 (Dibbens et al., 2008). PCDH19 (protocadherin 19) has been identified as the second most common mutated gene in epilepsy after SCN1A, which leads to Dravet syndrome (Depienne & Leguern, 2012; Niazi et al., 2019; Perez et al., 2017). PCDH19 (OMIM: 300460) is located at Xq22.1 and comprises six exons that encode protocadherin-19, an 1148 amino-acid protein. It contains six extracellular cadherins (EC) domains with conserved calcium-binding sequences, a transmembrane domain, and a cytoplasmic domain (Hulpiau & Van Roy, 2009). Most pathological variants occur within the first and the largest exon, encoding the six EC domains and a transmembrane domain. Exons 2–6 encode the intracellular domain of the protein (Depienne & Leguern, 2012; Duszyc et al., 2015; Lyons et al., 2017). This protein belongs to the non-clustered (delta 2) protocadherin subclass of the cadherin superfamily, which is greatly expressed in nerve tissues and at various developmental periods (Dibbens et al., 2008; Gaitan & Bouchard, 2006; Wolverton & Lalande, 2001). PCDH19 partakes in calcium-dependent cell-to-cell adhesion, regulating neuronal connection and signal transduction in the early phases of neurodevelopment (Dibbens et al., 2008). Besides, during the early stages of postnatal life, PCDH19 contributes to the development of synaptic connections and the maintenance of synaptic connections during adulthood (Kim et al., 2010). This protein seems to regulate gamma-aminobutyric acid type A receptors (GABAARs). This adhesion molecule mediates the differentiation of neuronal progenitors, neuronal migration, and maturation by inducing GABAergic signaling (Bassani et al., 2018).
In recent years, a growing number of female and male patients with PCDH19-related epilepsy and symptoms have been reported. Here, we report two additional female patients with DEE9 who are siblings, and both have a heterozygous frameshift variant in the PCDH19 gene. Although there is a report of PCDH19 polymorphism in the Iranian population (Asadi et al., 2022), there is no report of disease-causing variants. This research is the first report of PCDH19 mutation in the Iranian population. We have also expanded the phenotypic spectrum of DEE9. Moreover, this report on parental germline mosaicism emphasizes the clinical importance of considering prenatal diagnosis (PND) in X-linked/autosomal dominant disorders.
2. Materials and Methods
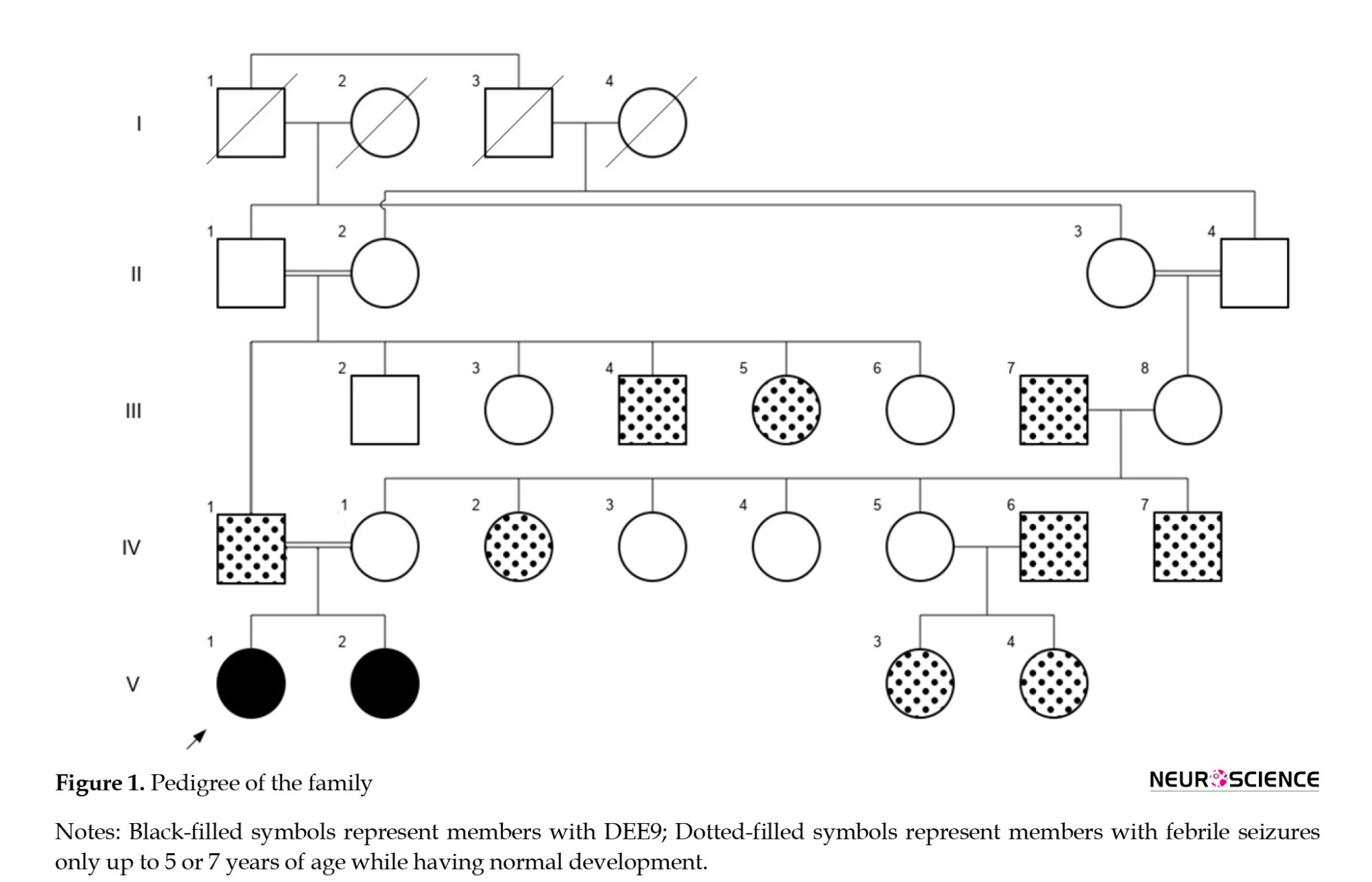
Here, we describe two female patients with DEE9 who are siblings. Their developmental milestones in motor (sitting, neck holding, rolling over, and walking) and social interactions (first social smile and responding to name) were normal for their age. After seizure onset, they showed developmental regression and some other conditions. Karyotype analyses were normal. Their perinatal history, pregnancy, and birth history were all unremarkable. There is a history of febrile seizures in some relatives with normal development (Figure 1). There was no family history of recurrent abortions, congenital anomalies, intellectual disability, or genetic disorders.
Case 1
The patient is a 5-year-old girl. She is the first child of consanguineous parents, from a 21-year-old mother and a 22-year-old father. She was born at full-term via normal vaginal delivery with a 2.5 kg birth weight. At the age of 5.5 months, she presented afebrile tonic seizures with lateral gaze lasting for 15-20 seconds and repeated 15 times a day. Three days later, a second seizure happened with identical characteristics, and then she had seizures every 2 months. At 14 months, she had recurrent seizures (4-5 times a week). At 1 year and 3 months of age, she exhibited speech regression and behavioral changes, including irritability, excessive crying, screaming, and attentional problems. At 2 years and 9 months of age, she had afebrile cluster seizures, which were mixed types, including tonic with lateral gaze, tonic-clonic, and myoclonic. Now, she still has refractory seizures, hypertelorism, hypoplastic midface, normal motor development, intellectual disability, impaired speech development, aggressive behavior, and autistic features (avoiding eye contact, impaired cognitive and learning skills, inattentive behavior, unresponsive to smile, getting very upset if she does not like certain clothing or sound), and constipation. Her current seizures are clustered, afebrile, and mostly tonic with lateral gaze. Brain magnetic resonance imaging (MRI) was unremarkable. The electroencephalography (EEG) analysis showed a normal background without asymmetry between hemispheres and some interictal epileptiform discharges (IEDs) in the right temporal region. Her seizures were partially controlled with anti-seizure medications (ASMs). Her previous medication regimen includes sodium valproate, nitrazepam, carbamazepine, levetiracetam, primidone, topiramate, and phenobarbital. The last ASM regimen includes topiramate and lacosamide, but she still suffers from recurrent seizures approximately every 2 months.
Case 2
The patient is the second child of consanguineous parents, from a 24-year-old mother and a 25-year-old father. She is a 2.5-year-old girl and was born at full-term via normal vaginal delivery. Birth weight was 2.55 kg, length was 48 cm, and head circumference at birth was 33 cm. At 6 months of age, she had febrile seizures provoked by vaccination, described as tonic seizures with lateral gaze lasting 20-25 seconds and repeated 3-4 times a day. Now, her seizures are well controlled with medication. She has normal motor development, mild intellectual disability, and autistic features (repetitive movement, hyperactivity, and delayed speech and language skills). Brain computed tomography (CT) scan was normal, and EEG showed a normal background without any asymmetry between hemispheres and some IEDs as bilaterally spikes in the occipital regions.
She has been on various ASMs. Her first episodes of seizures at 6 months of age were well controlled by levetiracetam and phenobarbital. At approximately 7 months, she had adverse drug reactions, and the ASM was switched to clobazam. After a seizure-free period lasting 5 months, seizures recurred at age 1 year due to arbitrarily discontinuing clobazam. Her next drug regimen included levetiracetam, topiramate, and phenytoin. Now, her seizures are controlled by the three ASMs: Levetiracetam, oxcarbazepine, and clobazam.
Study procedure
Participants, whole-exome sequencing (WES), bioinformatics analysis, and Sanger sequencing
Peripheral blood samples were collected from the proband, her sister, and parents after informed consent. Genomic DNA from blood samples was extracted using a Zistagen DNA extraction kit according to the manufacturer’s instructions. For the proband, WES was performed by capturing the targeted regions using Agilent SureSelect XT2 V7 (per manufacturer’s instructions). Then, sequencing was performed on the Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA) with 100 bp paired-end reads at an average sequencing depth 100×. Sequence reads were aligned to the GRCh37/hg19 human genome assembly using Burrows-Wheeler Aligner (BWA). Variant calling was done using SAMTools and Genome Analysis Toolkit (GATK v 3.7) (Li & Durbin, 2010; Li et al., 2009; McKenna et al., 2010). In addition, ANNOVAR software annotated and filtered the variants. For more filtrations, all pathogenic variants described in HGMD, and also variants with minor allele frequency (MAF) less than 0.01% were measured against gnomAD, ExAC, 1000 Genome project, dbSNP138, ESP6500, NHBL Exome Variant Server (EVS), and Iranome. The biological effects of candidate variant genes were determined in silico by predictor tools, including MutationTaster, SIFT, Polyphen 2, and CADD software. Eventually, sequencing result was also filtered based on phenotype, inheritance pattern, and variant type. Then, Sanger sequencing was carried out using ABI Prism3500 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). The primer sequences for PCR in Sanger sequencing are listed as follows: Forward primer, 5′- GGTGAGCGTGCCAGAAAAC -3′ and reverse primer, 5′- GTCAGTGATGAGCACGGTAAAG -3′.
Search strategy
The literature were searched using keywords and boolean operators “PCDH19” OR “Protocadherin-19” OR “KIAA1313” AND “mosaicism” in PubMed (until December 20, 2022). We included only English articles. A total of 41 potentially relevant records were found and screened: 11 records were cell line or animal studies. Then, 8 records were excluded because they were review articles. We also searched for previous reports of cases with c.1091delC (NM_001184880.2) in PCDH19, and 3 records were identified. Finally, 36 articles were selected for detailed study.
3. Results
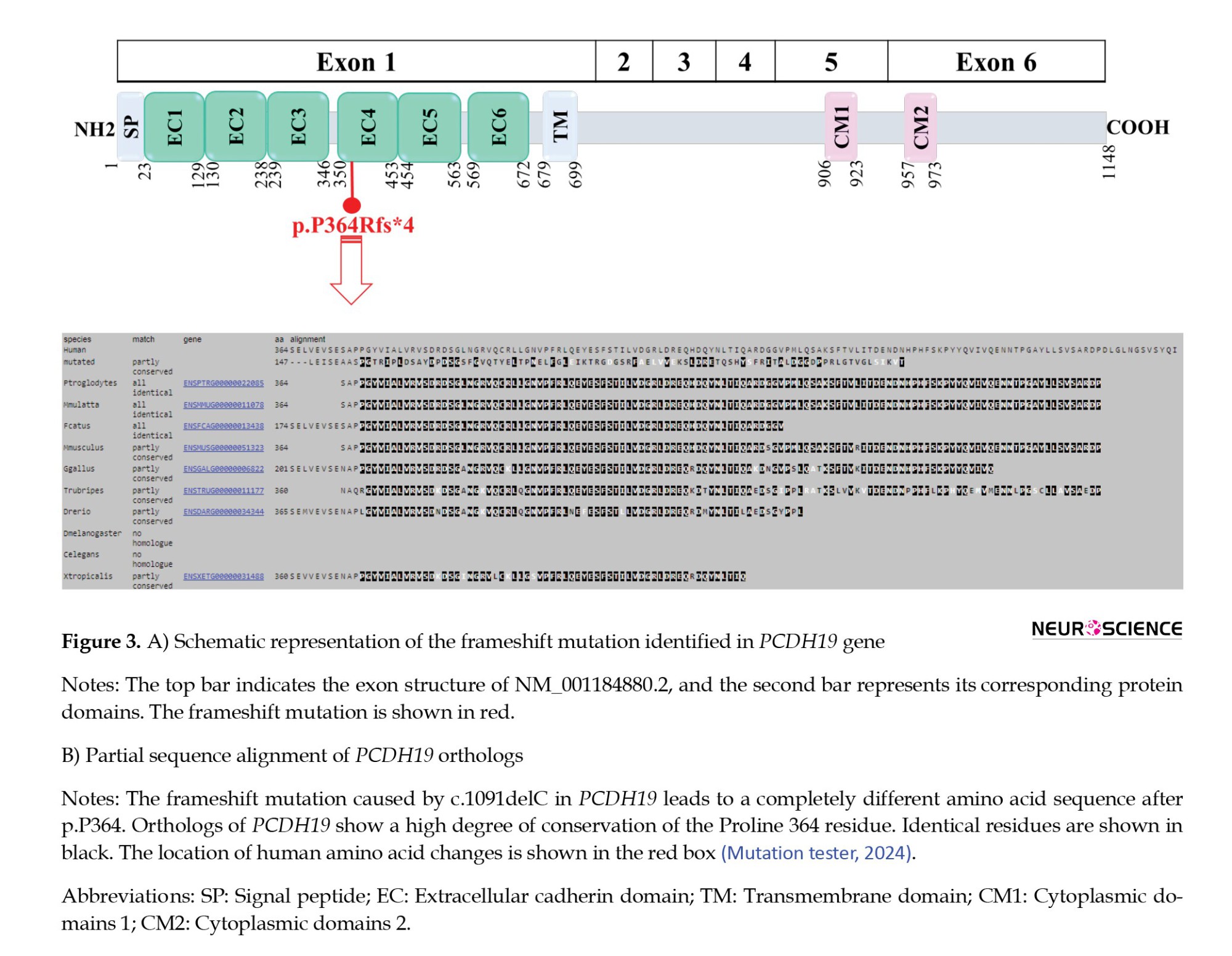
In consistency with the phenotype, the proband was detected to be heterozygous for c.1091delC (p.P364Rfs*4) in PCDH19 (RefSeq accession numbers NM_001184880.2 and NP_001171809.1). The number of alternate alleles/total read depth at this base position was 177/424 reads. This mutation was confirmed by Sanger sequencing, and a segregation study was carried out. The proband and her sister were found to be heterozygous for the variant, but it was not detected in the parents; therefore, it could be due to germline mosaicism (Figure 2). In this variation, a cytosine at coding position 1091 was deleted, and the mutation subsequently resulted in the change of amino acid at position 364 and ending in the fourth amino acid after the change (p.Pro364ArgfsTer4). Indeed, the variant disrupts the open reading frame and creates a premature stop codon in the mRNA of PCDH19. This condition presumably produces degradation of the mRNA in affected cells. According to the American College of Medical Genetics and Genomics (ACMG) guide (Richards et al., 2015), this gene mutation was considered pathogenic (PVS1 + PP5 + PM2). It w:::::::::as char:::::::::acterized as disease-causing in MutationTaster prediction tool. It was also reported as pathogenic in ClinVar. At the time of reporting, the variant does not exist in controls from gnomAD Aggregated and has a frequency of <0.01% in the ExAC database.

4. Discussion
With the broad use of next-generation sequencing technologies, numerous genes have been increasingly identified as the etiology of epilepsy. PCDH19 is the second most common genetic cause of epilepsy (Duszyc et al., 2015; Symonds & McTague, 2020). Pathogenic variants in PCDH19 lead to the development of epileptic encephalopathy 9. This disorder has extremely variable clinical manifestations, including early onset of different types and frequency of recurrent seizure clusters that are noticeably fever sensitive, mild to profound intellectual disability, autistic characteristics, and behavioral problems (Depienne & Leguern, 2012; Depienne et al., 2011; Higurashi et al., 2015; Perez et al., 2017; Steel et al., 2017; Trivisano et al., 2016; Van Harssel et al., 2013). The phenotypes of DEE9 and Dravet syndrome (OMIM #607208) can overlap; however, PCDH19-associated epilepsy has some specific features, such as later seizure onset, absence of photosensitivity, raised seizure cluster frequency, and satisfactory response to steroid treatment. Its inheritance is also distinct from the autosomal dominant inheritance in Dravet syndrome and selectively affects heterozygous females and mosaic hemizygous males (Depienne et al., 2009; Depienne & Leguern, 2012; Samanta, 2020). In DEE9 patients, the neuropsychiatric features in mosaic male patients resemble female phenotypes (De Lange et al., 2017; Kolc et al., 2020; Yang et al., 2020). In addition to the “cellular interference” hypothesis as a key pathogenic mechanism of DEE9 (Depienne et al., 2009; Depienne & Leguern, 2012), the other hypotheses also include decreased GABAA receptor function (Bassani et al., 2018; Serratto et al., 2020), allopregnanolone deficiency in females (Tan et al., 2015; Trivisano et al., 2017), and blood-brain barrier impairment (Higurashi et al., 2015). Recently, animal studies have been done to shed light on the related mechanisms. Lim et al. (2019) indicated that hemizygous Pcdh19 knockout (KO) male mice have autistic traits. So, additional studies will help to understand whether mosaic loss and complete loss of PCDH19 lead to autism-like phenotypes (Lim et al., 2019). Furthermore, Rakotomamonjy et al. (2020) investigated seizure susceptibility and progression in the Pchd19 mouse model. They observed increased susceptibility in Pcdh19 KO females, which proposes further mechanisms other than cellular interference have a role in PCDH19-associated epilepsy. The study implemented by Robens et al., (2022) in mosaic and non-mosaic pcdh19 mutant zebrafish challenged the theory that mosaicism is responsible for all PCDH19-related phenotypes. They suggested interneuron-mediated mechanisms govern these phenotypes (Robens et al., 2022). Despite these experiments, the exact mechanisms underlying the disorder are still unclear. Hence, further studies are required to understand and clarify the involved mechanisms.
In the pedigree depicted in Figure 1, multiple members with childhood febrile seizures lasted only up to 5 or 7 years of age without any medications, and all of them had normal development. The proband’s father also had this kind of seizure, but the variant detected in the proband was absent in her father. Therefore, other seizure susceptibility loci seem to be in this family, and further genetic studies should be performed on these individuals to identify these loci.
More than 270 variants of PCDH19 have been identified in female patients (Chen et al., 2022). The present study reported a frameshift mutation (c.1091delC, p.P364Rfs*4) in exon 1 of PCDH19 in two affected sisters, which occurred as germline mosaicism. Among all exons in PCDH19, exon 1 includes the highest number of pathogenic variants (Kolc et al., 2019; Wolverton & Lalande, 2001) and also has more pathogenic variants per base (5% per base) (Kolc et al., 2019). This condition suggests mutation in the extracellular domains of protocadherin-19 is poorly tolerated. Several studies on PCDH19 revealed that epilepsy onset time, mosaicism proportion, mutation types, and the affected domains are involved in the severity of encephalopathy (De Lange et al., 2017; Depienne et al., 2009; Kolc et al., 2019; Shibata et al., 2021). In this study, the detected frameshift mutation affects a highly conserved amino acid residue (PhastCons ≈ 1) in exon 1 at the EC 4 domain (UniProtKB- Q8TAB3), which contributes to calcium-dependent cell-cell interaction (Figure 3) (Depienne & Leguern, 2012; Niazi et al., 2019).
This variant had been previously reported in four female patients with epilepsy and neuropsychiatric manifestations (Table 1).

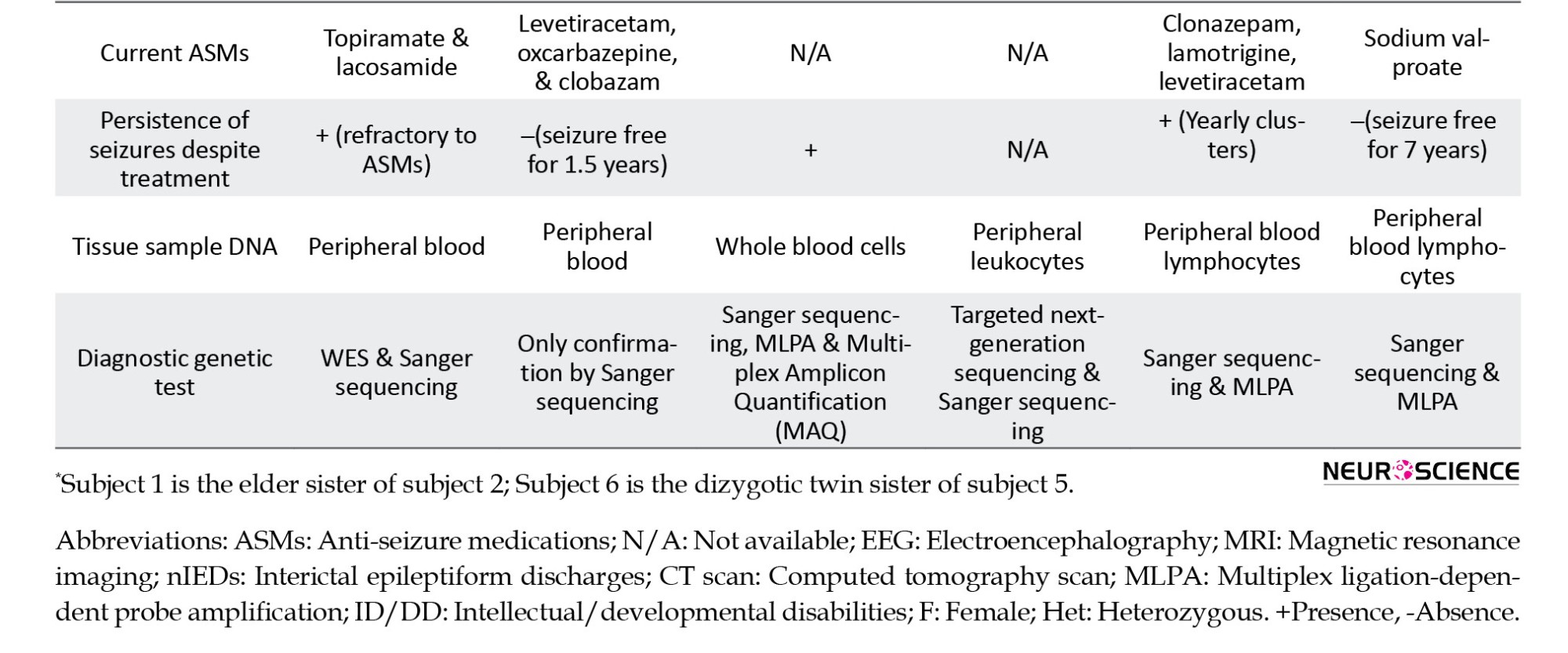
Our two cases, like the majority of DEE9 cases, had normal development before seizure onset, and therefore, speech delay/impairment, intellectual disability, and neuropsychiatric symptoms were observed after seizure onset. The 6 patients, summarized in Table 1, share some clinical characteristics, including early-onset seizures (5–18 months of age) and seizure clusters. In our cases (2 of 6 cases), the variant occurred as germline mosaicism and, in one case, as a de novo mutation; the remaining cases had paternal inheritance. In all patients, intellectual disability was detected, except for subject 6. Behavioral/psychiatric symptoms were found in 5 of 6 patients. Subject 5 had autistic features but was not observed in her sister with the same mutation. Moreover, despite taking appropriate doses of ASMs, subjects 1 and 5 had refractory seizures, but their sisters carrying the same mutation were seizure-free for 1.5 years and 7 years, respectively. In our 2 cases, although one sister had febrile seizures, the other did not experience any febrile seizures. Although dysmorphism and malformation are not commonly associated with PCDH19 (Depienne et al., 2011), we observed them in our proband. Indeed, dysmorphic features were only reported in our proband, but not in her sister and other reported cases with c.1091delC mutation. In addition to the intra-familial phenotypic heterogeneity detected in our 2 patients, it was also observed in subjects 5 and 6, who are dizygotic twins. Subjects 5 and 6 differ in some features, including age at seizure onset, seizure type, behavioral/psychiatric problems, intellectual disability, and response to ASMs (Liu et al., 2017). So, females with PCDH19 mutations display extensive intra-familial and inter-familial phenotypic variability. This diversity may be explained by different X-inactivation patterns in the brain, genetic modifiers, or mutation mosaicism in patients’ brains. This condition makes it difficult to predict the clinical course for unborn or young children (Scheffer et al., 2008). Females with heterozygous PCDH19 mutation have incomplete penetrance (approximately 90%) (Liu et al., 2019). Although epigenetic mechanisms, particularly skewed X- inactivation, are expected to have a pivotal role, there is limited data about the effect of X-chromosome inactivation on the penetrance of DEE9 (Hung et al., 2021).
There are also some other reports of PCDH19 mutation recurrence in a family. Dibbens et al. (2011) reported two unrelated families with two affected sisters. They demonstrated that germline mosaicism of a PCDH19 mutation in a parent is a crucial mechanism associated with EFMR inheritance (Dibbens et al., 2011). Furthermore, mosaicism of PCDH19 mutations has been found in mildly affected or unaffected mothers (Dibbens et al., 2011; Terracciano et al., 2012). This issue can be clarified by the random X-inactivation or the amount of mutant against wild-type protocadherin 19. Interestingly, Liu et al., (2017) reported an asymptomatic mosaic father with two affected girls (Liu et al., 2017). De novo mutations can be transmitted from mosaic parents (Xu et al., 2015). Considering asymptomatic mosaic fathers, it is proposed that the frequency of parental mosaicism is underestimated. Stosser et al. (2018) conducted a study investigating the frequency of mosaicism identified by next-generation sequencing in genes related to epilepsy-associated neurodevelopmental disorders. Their results showed that PCDH19 and CDKL5 had the highest frequency of mosaicism for a pathogenic or likely pathogenic variant (Stosser et al., 2018). Nevertheless, because of the technical limitations, mutations in mosaic parents could remain undiagnosed. Therefore, when there is a mutation in such genes in a patient, we recommend that genetic testing and PND be considered for the next family pregnancies.
Negi et al. (2023) conducted a study investigating the genetic explanation of diphtheria, tetanus, and whole-cell pertussis (DTwP) vaccination-associated seizures or subsequent epilepsies. They performed genetic testing on 54 children with DTwP vaccination-associated seizures. Their study revealed 33 pathogenic variants in 12 genes, with one variant detected in the PCDH19 gene (Negi et al., 2023). Whole-cell pertussis (wP) vaccination is also used in Iran, increasing the probability of vaccine-associated seizure and encephalopathy in individuals with a favorable genetic background. Moreover, in one of our cases with PCDH19 mutation, seizures started following DPwT vaccination at age 6 months. Hence, regarding these issues and the importance of diagnosis, prognosis, and potential individualized treatment, we recommend using next-generation sequencing in any child with vaccine-associated seizures. We also recommend educating healthcare professionals about the adverse effects of DPT vaccination.
There is no specific pharmacological treatment for PCDH19-associated epilepsy (Moncayo et al., 2022). Therefore, a better understanding of the pathogenic mechanisms underlying DEE9 would pave the way for developing mechanism-based approaches. An important step for optimizing these precision therapies is having more knowledge about PCDH19 genetic variants and the biological processes disrupted by these mutations (Dell’Isola et al., 2022). For example, modifying GABAergic activity would be an effective therapy if the inhibitory neurons are reduced. With precision therapies, there is hope that the prognosis of patients will improve in the future. Moreover, there is an immense need to perform research on novel advanced precision medicine approaches, including different gene therapy strategies. These novel approaches will potentially provide effective treatment for this rare genetic disorder in the future (Bertocchi et al., 2023).
In genetic epilepsies such as DEEs, diagnosis of a specific genetic defect could have multiple benefits for the patients and their families. Confirming a molecular etiology can help the patient choose the most appropriate treatment. In particular types of DEEs, there are targeted therapies that can improve seizure burden and neurodevelopmental outcomes in the affected children. In addition, molecular diagnosis will also impact the genetic counseling of the patients and families and make them aware of the prognosis, recurrence risk, reproductive options, and PND. Furthermore, a specific genetic diagnosis provides peace of mind and reduces the need for further diagnostic tests (Syrbe, 2022). We recommend considering genetic testing in the care of children with DEEs as a routine diagnostic approach.
5. Conclusion
Our research is the first report of germline mosaicism in the PCDH19 gene in the Iranian population. Our study expanded the phenotypic spectrum of PCDH19-related developmental and epileptic encephalopathy (DEE9). Genetic testing has become an effective way of determining the diagnosis. Parental germline mosaicism should be taken into account when providing genetic counseling for X-linked/autosomal dominant disorders. This report emphasizes the importance of considering PND in such cases.
Compliance with ethical guidelines
All ethical principles are considered in this article. The patients’ parents had written informed consent to publish this information.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
Conceptualization, supervision and funding administration: Mohammad Miryounesi; Methodology: Sahar Alijanpour and amareh Panjeshahi; Investigation, data analysis and writing the original draft: Sahar Alijanpour;Data collection: Seyed Hassan Tonekaboni, Parvaneh Karimzadeh, Farzad Ahmadabadi, Elham Rahimian; Review and editing: Soudeh Ghafouri-Fard, Mohammad Miryounesi; Final approval: All authors.
Conflict of interest
The authors declared no conflict of interest.
Acknowledgments
The authors thank the children’s parents for their collaboration.
References
Epilepsy is one of the most common neurological conditions among children, and its highest incidence is seen in the first year of life (Fine & Wirrell, 2020). This is a heterogeneous group of disorders characterized by the triad of early-onset epileptic seizures, abnormal EEG activity, and developmental impairment, which are called developmental and epileptic encephalopathies (DEEs). They may be caused by both genetic and non-genetic etiologies (Guerrini et al., 2023). We have different types of DEEs, and one of them is developmental and epileptic encephalopathy 9 (DEE9), which results from pathogenic variants in the PCDH19 gene (Kolc et al., 2019). Several names have been adopted to signify the clinical characterization of this disorder, including girls clustering epilepsy (PCDH19- GCE) (Kolc et al., 2019), ‘‘early infantile epileptic encephalopathy-9” (EIE19) or developmental and epileptic encephalopathy 9 (DEE9), and PCDH19 clustering epilepsy (PCDH19-CE) (Depienne et al., 2009; Kolc et al., 2020). Here, we use DEE9 (OMIM #300088) to refer to the disorder.
Symptoms of DEE9 patients evolve in early infancy, mostly within the first year of life. It is often provoked by fever and presents clustered seizures, different degrees of cognitive impairment, and behavioral problems, such as autism spectrum disorder (ASD), hyperactivity, attention deficit, and aggression (Kolc et al., 2020). The clinical characteristics may be similar to those of Dravet syndrome (Depienne et al., 2009). The disorder is defined by a distinctive X-linked inheritance pattern where heterozygous females or rarely mosaic hemizygous males are affected, but hemizygous males and homozygous females are asymptomatic. PCDH19 is located in the region exposed to X-chromosome inactivation. Random X-inactivation and consequent tissue mosaicism are posited to be implicated in the pathogenesis of heterozygous females (Depienne et al., 2009; Depienne & Leguern, 2012). Although the exact underlying mechanism is still unclear, cellular interference has been proposed to clarify this unusual inheritance pattern (Depienne et al., 2009; Depienne & Leguern, 2012; Samanta, 2020; Thiffault et al., 2016). The cellular interference hypothesis postulates that the existence of a variety of cells in the brain, expressing either mutant or wild-type PCDH19 protein, results in abnormal neurodevelopment and is therefore the cardinal cause of the clinical presentations (Depienne et al., 2009).
The causative gene of DEE9 was mapped to the X chromosome in 1997 (Ryan et al., 1997) and determined to be PCDH19 in 2008 (Dibbens et al., 2008). PCDH19 (protocadherin 19) has been identified as the second most common mutated gene in epilepsy after SCN1A, which leads to Dravet syndrome (Depienne & Leguern, 2012; Niazi et al., 2019; Perez et al., 2017). PCDH19 (OMIM: 300460) is located at Xq22.1 and comprises six exons that encode protocadherin-19, an 1148 amino-acid protein. It contains six extracellular cadherins (EC) domains with conserved calcium-binding sequences, a transmembrane domain, and a cytoplasmic domain (Hulpiau & Van Roy, 2009). Most pathological variants occur within the first and the largest exon, encoding the six EC domains and a transmembrane domain. Exons 2–6 encode the intracellular domain of the protein (Depienne & Leguern, 2012; Duszyc et al., 2015; Lyons et al., 2017). This protein belongs to the non-clustered (delta 2) protocadherin subclass of the cadherin superfamily, which is greatly expressed in nerve tissues and at various developmental periods (Dibbens et al., 2008; Gaitan & Bouchard, 2006; Wolverton & Lalande, 2001). PCDH19 partakes in calcium-dependent cell-to-cell adhesion, regulating neuronal connection and signal transduction in the early phases of neurodevelopment (Dibbens et al., 2008). Besides, during the early stages of postnatal life, PCDH19 contributes to the development of synaptic connections and the maintenance of synaptic connections during adulthood (Kim et al., 2010). This protein seems to regulate gamma-aminobutyric acid type A receptors (GABAARs). This adhesion molecule mediates the differentiation of neuronal progenitors, neuronal migration, and maturation by inducing GABAergic signaling (Bassani et al., 2018).
In recent years, a growing number of female and male patients with PCDH19-related epilepsy and symptoms have been reported. Here, we report two additional female patients with DEE9 who are siblings, and both have a heterozygous frameshift variant in the PCDH19 gene. Although there is a report of PCDH19 polymorphism in the Iranian population (Asadi et al., 2022), there is no report of disease-causing variants. This research is the first report of PCDH19 mutation in the Iranian population. We have also expanded the phenotypic spectrum of DEE9. Moreover, this report on parental germline mosaicism emphasizes the clinical importance of considering prenatal diagnosis (PND) in X-linked/autosomal dominant disorders.
2. Materials and Methods
Here, we describe two female patients with DEE9 who are siblings. Their developmental milestones in motor (sitting, neck holding, rolling over, and walking) and social interactions (first social smile and responding to name) were normal for their age. After seizure onset, they showed developmental regression and some other conditions. Karyotype analyses were normal. Their perinatal history, pregnancy, and birth history were all unremarkable. There is a history of febrile seizures in some relatives with normal development (Figure 1). There was no family history of recurrent abortions, congenital anomalies, intellectual disability, or genetic disorders.
Case 1
The patient is a 5-year-old girl. She is the first child of consanguineous parents, from a 21-year-old mother and a 22-year-old father. She was born at full-term via normal vaginal delivery with a 2.5 kg birth weight. At the age of 5.5 months, she presented afebrile tonic seizures with lateral gaze lasting for 15-20 seconds and repeated 15 times a day. Three days later, a second seizure happened with identical characteristics, and then she had seizures every 2 months. At 14 months, she had recurrent seizures (4-5 times a week). At 1 year and 3 months of age, she exhibited speech regression and behavioral changes, including irritability, excessive crying, screaming, and attentional problems. At 2 years and 9 months of age, she had afebrile cluster seizures, which were mixed types, including tonic with lateral gaze, tonic-clonic, and myoclonic. Now, she still has refractory seizures, hypertelorism, hypoplastic midface, normal motor development, intellectual disability, impaired speech development, aggressive behavior, and autistic features (avoiding eye contact, impaired cognitive and learning skills, inattentive behavior, unresponsive to smile, getting very upset if she does not like certain clothing or sound), and constipation. Her current seizures are clustered, afebrile, and mostly tonic with lateral gaze. Brain magnetic resonance imaging (MRI) was unremarkable. The electroencephalography (EEG) analysis showed a normal background without asymmetry between hemispheres and some interictal epileptiform discharges (IEDs) in the right temporal region. Her seizures were partially controlled with anti-seizure medications (ASMs). Her previous medication regimen includes sodium valproate, nitrazepam, carbamazepine, levetiracetam, primidone, topiramate, and phenobarbital. The last ASM regimen includes topiramate and lacosamide, but she still suffers from recurrent seizures approximately every 2 months.
Case 2
The patient is the second child of consanguineous parents, from a 24-year-old mother and a 25-year-old father. She is a 2.5-year-old girl and was born at full-term via normal vaginal delivery. Birth weight was 2.55 kg, length was 48 cm, and head circumference at birth was 33 cm. At 6 months of age, she had febrile seizures provoked by vaccination, described as tonic seizures with lateral gaze lasting 20-25 seconds and repeated 3-4 times a day. Now, her seizures are well controlled with medication. She has normal motor development, mild intellectual disability, and autistic features (repetitive movement, hyperactivity, and delayed speech and language skills). Brain computed tomography (CT) scan was normal, and EEG showed a normal background without any asymmetry between hemispheres and some IEDs as bilaterally spikes in the occipital regions.
She has been on various ASMs. Her first episodes of seizures at 6 months of age were well controlled by levetiracetam and phenobarbital. At approximately 7 months, she had adverse drug reactions, and the ASM was switched to clobazam. After a seizure-free period lasting 5 months, seizures recurred at age 1 year due to arbitrarily discontinuing clobazam. Her next drug regimen included levetiracetam, topiramate, and phenytoin. Now, her seizures are controlled by the three ASMs: Levetiracetam, oxcarbazepine, and clobazam.
Study procedure
Participants, whole-exome sequencing (WES), bioinformatics analysis, and Sanger sequencing
Peripheral blood samples were collected from the proband, her sister, and parents after informed consent. Genomic DNA from blood samples was extracted using a Zistagen DNA extraction kit according to the manufacturer’s instructions. For the proband, WES was performed by capturing the targeted regions using Agilent SureSelect XT2 V7 (per manufacturer’s instructions). Then, sequencing was performed on the Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA) with 100 bp paired-end reads at an average sequencing depth 100×. Sequence reads were aligned to the GRCh37/hg19 human genome assembly using Burrows-Wheeler Aligner (BWA). Variant calling was done using SAMTools and Genome Analysis Toolkit (GATK v 3.7) (Li & Durbin, 2010; Li et al., 2009; McKenna et al., 2010). In addition, ANNOVAR software annotated and filtered the variants. For more filtrations, all pathogenic variants described in HGMD, and also variants with minor allele frequency (MAF) less than 0.01% were measured against gnomAD, ExAC, 1000 Genome project, dbSNP138, ESP6500, NHBL Exome Variant Server (EVS), and Iranome. The biological effects of candidate variant genes were determined in silico by predictor tools, including MutationTaster, SIFT, Polyphen 2, and CADD software. Eventually, sequencing result was also filtered based on phenotype, inheritance pattern, and variant type. Then, Sanger sequencing was carried out using ABI Prism3500 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). The primer sequences for PCR in Sanger sequencing are listed as follows: Forward primer, 5′- GGTGAGCGTGCCAGAAAAC -3′ and reverse primer, 5′- GTCAGTGATGAGCACGGTAAAG -3′.
Search strategy
The literature were searched using keywords and boolean operators “PCDH19” OR “Protocadherin-19” OR “KIAA1313” AND “mosaicism” in PubMed (until December 20, 2022). We included only English articles. A total of 41 potentially relevant records were found and screened: 11 records were cell line or animal studies. Then, 8 records were excluded because they were review articles. We also searched for previous reports of cases with c.1091delC (NM_001184880.2) in PCDH19, and 3 records were identified. Finally, 36 articles were selected for detailed study.
3. Results
In consistency with the phenotype, the proband was detected to be heterozygous for c.1091delC (p.P364Rfs*4) in PCDH19 (RefSeq accession numbers NM_001184880.2 and NP_001171809.1). The number of alternate alleles/total read depth at this base position was 177/424 reads. This mutation was confirmed by Sanger sequencing, and a segregation study was carried out. The proband and her sister were found to be heterozygous for the variant, but it was not detected in the parents; therefore, it could be due to germline mosaicism (Figure 2). In this variation, a cytosine at coding position 1091 was deleted, and the mutation subsequently resulted in the change of amino acid at position 364 and ending in the fourth amino acid after the change (p.Pro364ArgfsTer4). Indeed, the variant disrupts the open reading frame and creates a premature stop codon in the mRNA of PCDH19. This condition presumably produces degradation of the mRNA in affected cells. According to the American College of Medical Genetics and Genomics (ACMG) guide (Richards et al., 2015), this gene mutation was considered pathogenic (PVS1 + PP5 + PM2). It w:::::::::as char:::::::::acterized as disease-causing in MutationTaster prediction tool. It was also reported as pathogenic in ClinVar. At the time of reporting, the variant does not exist in controls from gnomAD Aggregated and has a frequency of <0.01% in the ExAC database.
4. Discussion
With the broad use of next-generation sequencing technologies, numerous genes have been increasingly identified as the etiology of epilepsy. PCDH19 is the second most common genetic cause of epilepsy (Duszyc et al., 2015; Symonds & McTague, 2020). Pathogenic variants in PCDH19 lead to the development of epileptic encephalopathy 9. This disorder has extremely variable clinical manifestations, including early onset of different types and frequency of recurrent seizure clusters that are noticeably fever sensitive, mild to profound intellectual disability, autistic characteristics, and behavioral problems (Depienne & Leguern, 2012; Depienne et al., 2011; Higurashi et al., 2015; Perez et al., 2017; Steel et al., 2017; Trivisano et al., 2016; Van Harssel et al., 2013). The phenotypes of DEE9 and Dravet syndrome (OMIM #607208) can overlap; however, PCDH19-associated epilepsy has some specific features, such as later seizure onset, absence of photosensitivity, raised seizure cluster frequency, and satisfactory response to steroid treatment. Its inheritance is also distinct from the autosomal dominant inheritance in Dravet syndrome and selectively affects heterozygous females and mosaic hemizygous males (Depienne et al., 2009; Depienne & Leguern, 2012; Samanta, 2020). In DEE9 patients, the neuropsychiatric features in mosaic male patients resemble female phenotypes (De Lange et al., 2017; Kolc et al., 2020; Yang et al., 2020). In addition to the “cellular interference” hypothesis as a key pathogenic mechanism of DEE9 (Depienne et al., 2009; Depienne & Leguern, 2012), the other hypotheses also include decreased GABAA receptor function (Bassani et al., 2018; Serratto et al., 2020), allopregnanolone deficiency in females (Tan et al., 2015; Trivisano et al., 2017), and blood-brain barrier impairment (Higurashi et al., 2015). Recently, animal studies have been done to shed light on the related mechanisms. Lim et al. (2019) indicated that hemizygous Pcdh19 knockout (KO) male mice have autistic traits. So, additional studies will help to understand whether mosaic loss and complete loss of PCDH19 lead to autism-like phenotypes (Lim et al., 2019). Furthermore, Rakotomamonjy et al. (2020) investigated seizure susceptibility and progression in the Pchd19 mouse model. They observed increased susceptibility in Pcdh19 KO females, which proposes further mechanisms other than cellular interference have a role in PCDH19-associated epilepsy. The study implemented by Robens et al., (2022) in mosaic and non-mosaic pcdh19 mutant zebrafish challenged the theory that mosaicism is responsible for all PCDH19-related phenotypes. They suggested interneuron-mediated mechanisms govern these phenotypes (Robens et al., 2022). Despite these experiments, the exact mechanisms underlying the disorder are still unclear. Hence, further studies are required to understand and clarify the involved mechanisms.
In the pedigree depicted in Figure 1, multiple members with childhood febrile seizures lasted only up to 5 or 7 years of age without any medications, and all of them had normal development. The proband’s father also had this kind of seizure, but the variant detected in the proband was absent in her father. Therefore, other seizure susceptibility loci seem to be in this family, and further genetic studies should be performed on these individuals to identify these loci.
More than 270 variants of PCDH19 have been identified in female patients (Chen et al., 2022). The present study reported a frameshift mutation (c.1091delC, p.P364Rfs*4) in exon 1 of PCDH19 in two affected sisters, which occurred as germline mosaicism. Among all exons in PCDH19, exon 1 includes the highest number of pathogenic variants (Kolc et al., 2019; Wolverton & Lalande, 2001) and also has more pathogenic variants per base (5% per base) (Kolc et al., 2019). This condition suggests mutation in the extracellular domains of protocadherin-19 is poorly tolerated. Several studies on PCDH19 revealed that epilepsy onset time, mosaicism proportion, mutation types, and the affected domains are involved in the severity of encephalopathy (De Lange et al., 2017; Depienne et al., 2009; Kolc et al., 2019; Shibata et al., 2021). In this study, the detected frameshift mutation affects a highly conserved amino acid residue (PhastCons ≈ 1) in exon 1 at the EC 4 domain (UniProtKB- Q8TAB3), which contributes to calcium-dependent cell-cell interaction (Figure 3) (Depienne & Leguern, 2012; Niazi et al., 2019).
This variant had been previously reported in four female patients with epilepsy and neuropsychiatric manifestations (Table 1).
Our two cases, like the majority of DEE9 cases, had normal development before seizure onset, and therefore, speech delay/impairment, intellectual disability, and neuropsychiatric symptoms were observed after seizure onset. The 6 patients, summarized in Table 1, share some clinical characteristics, including early-onset seizures (5–18 months of age) and seizure clusters. In our cases (2 of 6 cases), the variant occurred as germline mosaicism and, in one case, as a de novo mutation; the remaining cases had paternal inheritance. In all patients, intellectual disability was detected, except for subject 6. Behavioral/psychiatric symptoms were found in 5 of 6 patients. Subject 5 had autistic features but was not observed in her sister with the same mutation. Moreover, despite taking appropriate doses of ASMs, subjects 1 and 5 had refractory seizures, but their sisters carrying the same mutation were seizure-free for 1.5 years and 7 years, respectively. In our 2 cases, although one sister had febrile seizures, the other did not experience any febrile seizures. Although dysmorphism and malformation are not commonly associated with PCDH19 (Depienne et al., 2011), we observed them in our proband. Indeed, dysmorphic features were only reported in our proband, but not in her sister and other reported cases with c.1091delC mutation. In addition to the intra-familial phenotypic heterogeneity detected in our 2 patients, it was also observed in subjects 5 and 6, who are dizygotic twins. Subjects 5 and 6 differ in some features, including age at seizure onset, seizure type, behavioral/psychiatric problems, intellectual disability, and response to ASMs (Liu et al., 2017). So, females with PCDH19 mutations display extensive intra-familial and inter-familial phenotypic variability. This diversity may be explained by different X-inactivation patterns in the brain, genetic modifiers, or mutation mosaicism in patients’ brains. This condition makes it difficult to predict the clinical course for unborn or young children (Scheffer et al., 2008). Females with heterozygous PCDH19 mutation have incomplete penetrance (approximately 90%) (Liu et al., 2019). Although epigenetic mechanisms, particularly skewed X- inactivation, are expected to have a pivotal role, there is limited data about the effect of X-chromosome inactivation on the penetrance of DEE9 (Hung et al., 2021).
There are also some other reports of PCDH19 mutation recurrence in a family. Dibbens et al. (2011) reported two unrelated families with two affected sisters. They demonstrated that germline mosaicism of a PCDH19 mutation in a parent is a crucial mechanism associated with EFMR inheritance (Dibbens et al., 2011). Furthermore, mosaicism of PCDH19 mutations has been found in mildly affected or unaffected mothers (Dibbens et al., 2011; Terracciano et al., 2012). This issue can be clarified by the random X-inactivation or the amount of mutant against wild-type protocadherin 19. Interestingly, Liu et al., (2017) reported an asymptomatic mosaic father with two affected girls (Liu et al., 2017). De novo mutations can be transmitted from mosaic parents (Xu et al., 2015). Considering asymptomatic mosaic fathers, it is proposed that the frequency of parental mosaicism is underestimated. Stosser et al. (2018) conducted a study investigating the frequency of mosaicism identified by next-generation sequencing in genes related to epilepsy-associated neurodevelopmental disorders. Their results showed that PCDH19 and CDKL5 had the highest frequency of mosaicism for a pathogenic or likely pathogenic variant (Stosser et al., 2018). Nevertheless, because of the technical limitations, mutations in mosaic parents could remain undiagnosed. Therefore, when there is a mutation in such genes in a patient, we recommend that genetic testing and PND be considered for the next family pregnancies.
Negi et al. (2023) conducted a study investigating the genetic explanation of diphtheria, tetanus, and whole-cell pertussis (DTwP) vaccination-associated seizures or subsequent epilepsies. They performed genetic testing on 54 children with DTwP vaccination-associated seizures. Their study revealed 33 pathogenic variants in 12 genes, with one variant detected in the PCDH19 gene (Negi et al., 2023). Whole-cell pertussis (wP) vaccination is also used in Iran, increasing the probability of vaccine-associated seizure and encephalopathy in individuals with a favorable genetic background. Moreover, in one of our cases with PCDH19 mutation, seizures started following DPwT vaccination at age 6 months. Hence, regarding these issues and the importance of diagnosis, prognosis, and potential individualized treatment, we recommend using next-generation sequencing in any child with vaccine-associated seizures. We also recommend educating healthcare professionals about the adverse effects of DPT vaccination.
There is no specific pharmacological treatment for PCDH19-associated epilepsy (Moncayo et al., 2022). Therefore, a better understanding of the pathogenic mechanisms underlying DEE9 would pave the way for developing mechanism-based approaches. An important step for optimizing these precision therapies is having more knowledge about PCDH19 genetic variants and the biological processes disrupted by these mutations (Dell’Isola et al., 2022). For example, modifying GABAergic activity would be an effective therapy if the inhibitory neurons are reduced. With precision therapies, there is hope that the prognosis of patients will improve in the future. Moreover, there is an immense need to perform research on novel advanced precision medicine approaches, including different gene therapy strategies. These novel approaches will potentially provide effective treatment for this rare genetic disorder in the future (Bertocchi et al., 2023).
In genetic epilepsies such as DEEs, diagnosis of a specific genetic defect could have multiple benefits for the patients and their families. Confirming a molecular etiology can help the patient choose the most appropriate treatment. In particular types of DEEs, there are targeted therapies that can improve seizure burden and neurodevelopmental outcomes in the affected children. In addition, molecular diagnosis will also impact the genetic counseling of the patients and families and make them aware of the prognosis, recurrence risk, reproductive options, and PND. Furthermore, a specific genetic diagnosis provides peace of mind and reduces the need for further diagnostic tests (Syrbe, 2022). We recommend considering genetic testing in the care of children with DEEs as a routine diagnostic approach.
5. Conclusion
Our research is the first report of germline mosaicism in the PCDH19 gene in the Iranian population. Our study expanded the phenotypic spectrum of PCDH19-related developmental and epileptic encephalopathy (DEE9). Genetic testing has become an effective way of determining the diagnosis. Parental germline mosaicism should be taken into account when providing genetic counseling for X-linked/autosomal dominant disorders. This report emphasizes the importance of considering PND in such cases.
Compliance with ethical guidelines
All ethical principles are considered in this article. The patients’ parents had written informed consent to publish this information.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
Conceptualization, supervision and funding administration: Mohammad Miryounesi; Methodology: Sahar Alijanpour and amareh Panjeshahi; Investigation, data analysis and writing the original draft: Sahar Alijanpour;Data collection: Seyed Hassan Tonekaboni, Parvaneh Karimzadeh, Farzad Ahmadabadi, Elham Rahimian; Review and editing: Soudeh Ghafouri-Fard, Mohammad Miryounesi; Final approval: All authors.
Conflict of interest
The authors declared no conflict of interest.
Acknowledgments
The authors thank the children’s parents for their collaboration.
References
Asadi, F., Goodarzi, H., Zahiri, J., & Jaafarnia, M. (2022). [The identification of the disease -causing mutations in genes associated with episodic coma in a family with three girls affected with this disorder using Next Generation Sequencing (NGS)(Persian)]. Modares Journal of Biotechnology, 12(2), 67-74. [Link]
Bassani, S., Cwetsch, A. W., Gerosa, L., Serratto, G. M., Folci, A., & Hall, I. F., et al. (2018). The female epilepsy protein PCDH19 is a new GABAAR-binding partner that regulates GABAergic transmission as well as migration and morphological maturation of hippocampal neurons. Human Molecular Genetics, 27(6), 1027-1038. [DOI:10.1093/hmg/ddy019] [PMID]
Bertocchi, I., Cambiaghi, M., & Hasan, M. T. (2023). Advances toward precision therapeutics for developmental and epileptic encephalopathies. Frontiers in Neuroscience, 17, 1140679.[DOI:10.3389/fnins.2023.1140679] [PMID]
Chen, G., Zhou, H., Lu, Y., Wang, Y., Li, Y., & Xue, J., et al. (2022). Case report: A novel mosaic nonsense mutation of PCDH19 in a Chinese male with febrile epilepsy. Frontiers in Neurology, 13, 992781. [DOI:10.3389/fneur.2022.992781] [PMID]
de Lange, I. M., Rump, P., Neuteboom, R. F., Augustijn, P. B., Hodges, K., & Kistemaker, A. I., et al. (2017). Male patients affected by mosaic PCDH19 mutations: Five new cases. Neurogenetics, 18(3), 147-153. [DOI:10.1007/s10048-017-0517-5] [PMID]
Dell'Isola, G. B., Vinti, V., Fattorusso, A., Tascini, G., Mencaroni, E., & Di Cara, G., et al. (2022). The broad clinical spectrum of epilepsies associated with protocadherin 19 gene mutation. Frontiers in Neurology, 12, 780053. [DOI:10.3389/fneur.2021.780053] [PMID]
Depienne, C., Bouteiller, D., Keren, B., Cheuret, E., Poirier, K., & Trouillard, O., et al. (2009). Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. Plos Genetics, 5(2), e1000381. [DOI:10.1371/journal.pgen.1000381] [PMID]
Depienne, C., & Leguern, E. (2012). PCDH19‐related infantile epileptic encephalopathy: An unusual X‐linked inheritance disorder. Human Mutation, 33(4), 627-634. [DOI:10.1002/humu.22029] [PMID]
Depienne, C., Trouillard, O., Bouteiller, D., Gourfinkel-An, I., Poirier, K., & Rivier, F., et al. (2011). Mutations and deletions in PCDH19 account for various familial or isolated epilepsies in females. Human Mutation, 32(1), E1959-E1975. [DOI:10.1002/humu.21373] [PMID]
Dibbens, L. M., Kneen, R., Bayly, M. A., Heron, S. E., Arsov, T., & Damiano, J. A., et al. (2011). Recurrence risk of epilepsy and mental retardation in females due to parental mosaicism of PCDH19 mutations. Neurology, 76(17), 1514-1519. [DOI:10.1212/WNL.0b013e318217e7b6] [PMID]
Dibbens, L. M., Tarpey, P. S., Hynes, K., Bayly, M. A., Scheffer, I. E., & Smith, R., et al. (2008). X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nature Genetics, 40(6), 776-781. [DOI:10.1038/ng.149] [PMID]
Duszyc, K., Terczynska, I., & Hoffman-Zacharska, D. (2015). Epilepsy and mental retardation restricted to females: X-linked epileptic infantile encephalopathy of unusual inheritance. Journal of Applied Genetics, 56(1), 49-56. [DOI:10.1007/s13353-014-0243-8] [PMID]
Fine, A., & Wirrell, E. C. (2020). Seizures in children. Pediatrics in Review, 41(7), 321-347. [DOI:10.1542/pir.2019-0134] [PMID]
Gaitan, Y., & Bouchard, M. (2006). Expression of the δ-protocadherin gene Pcdh19 in the developing mouse embryo. Gene Expression Patterns, 6(8), 893-899. [DOI:10.1016/j.modgep.2006.03.001] [PMID]
Guerrini, R., Conti, V., Mantegazza, M., Balestrini, S., Galanopoulou, A. S., & Benfenati, F. (2023). Developmental and epileptic encephalopathies: From genetic heterogeneity to phenotypic continuum. Physiological Reviews, 103(1), 433-513. [DOI:10.1152/physrev.00063.2021] [PMID]
Higurashi, N., Takahashi, Y., Kashimada, A., Sugawara, Y., Sakuma, H., & Tomonoh, Y., et al. (2015). Immediate suppression of seizure clusters by corticosteroids in PCDH19 female epilepsy. Seizure, 27, 1-5. [DOI:10.1016/j.seizure.2015.02.006] [PMID]
Hulpiau, P., & Van Roy, F. (2009). Molecular evolution of the cadherin superfamily. The International Journal of Biochemistry & Cell Biology, 41(2), 349-369. [DOI:10.1016/j.biocel.2008.09.027] [PMID]
Hung, L. Y., Subramaniam, S. R., Tong, T. T., Chan, W. K., Yau, E. K., & Ching, C. K. (2021). X-chromosome inactivation and PCDH19-associated epileptic encephalopathy: A novel PCDH19 variant in a Chinese family. Clinica Chimica Acta, 521, 285-288. [DOI:10.1016/j.cca.2021.07.023] [PMID]
Kim, S. Y., Mo, J. W., Han, S., Choi, S. Y., Han, S. B., & Moon, B. H., et al. (2010). The expression of non-clustered protocadherins in adult rat hippocampal formation and the connecting brain regions. Neuroscience, 170(1), 189-199. [DOI:10.1016/j.neuroscience.2010.05.027] [PMID]
Kolc, K. L., Møller, R. S., Sadleir, L. G., Scheffer, I. E., Kumar, R., & Gecz, J. (2020). PCDH19 pathogenic variants in males: Expanding the phenotypic spectrum. Advances in Experimental Medicine and Biology, 1298, 177–187. [DOI:10.1007/5584_2020_574] [PMID]
Kolc, K. L., Sadleir, L. G., Scheffer, I. E., Ivancevic, A., Roberts, R., & Pham, D. H., et al. (2019). A systematic review and meta-analysis of 271 PCDH19-variant individuals identifies psychiatric comorbidities, and association of seizure onset and disease severity. Molecular Psychiatry, 24(2), 241-251. [DOI:10.1038/s41380-018-0066-9] [PMID]
Li, H., & Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics, 26(5), 589-595. [DOI:10.1093/bioinformatics/btp698] [PMID]
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., & Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics, 25(16), 2078-2079. [DOI:10.1093/bioinformatics/btp352] [PMID]
Lim, J., Ryu, J., Kang, S., Noh, H. J., & Kim, C. H. (2019). Autism-like behaviors in male mice with a Pcdh19 deletion. Molecular Brain, 12(1), 95. [DOI:10.1186/s13041-019-0519-3] [PMID]
Liu, A., Xu, X., Yang, X., Jiang, Y., Yang, Z., & Liu, X., et al. (2017). The clinical spectrum of female epilepsy patients with PCDH19 mutations in a Chinese population. Clinical Genetics, 91(1), 54-62. [DOI:10.1111/cge.12846] [PMID]
Liu, A., Yang, X., Yang, X., Wu, Q., Zhang, J., & Sun, D., (2019). Mosaicism and incomplete penetrance of PCDH19 mutations. Journal of Medical Genetics, 56(2), 81-88. [DOI:10.1136/jmedgenet-2017-105235] [PMID]
Lyons, S., Marnane, M., Reavey, E., Williams, N., & Costello, D. (2017). PCDH19-related epilepsy: A rare but recognisable clinical syndrome in females. Practical Neurology, 17(4), 314-317. [DOI:10.1136/practneurol-2016-001521] [PMID]
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., & Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research, 20(9), 1297-1303. [DOI:10.1101/gr.107524.110] [PMID]
Moncayo, J. A., Vargas, M. N., Castillo, I., Granda, P. V., Duque, A. M., & Argudo, J. M., et al. (2022). Adjuvant Treatment for Protocadherin 19 (PCDH19) Syndrome. Cureus, 14(7), e27154.[DOI:10.7759/cureus.27154] [PMID]
Negi, S., Sahu, J. K., Bhatia, P., Kaur, A., Malhi, P., & Singh, G., et al. (2023). Exploring the genetic landscape of diphtheria, tetanus and pertussis vaccination-associated seizures or subsequent epilepsies. The Lancet Regional Health-Southeast Asia, 8, 100094. [DOI:10.1016/j.lansea.2022.100094] [PMID]
Niazi, R., Fanning, E. A., Depienne, C., Sarmady, M., & Abou Tayoun, A. N. (2019). A mutation update for the PCDH19 gene causing early‐onset epilepsy in females with an unusual expression pattern. Human Mutation, 40(3), 243-257. [DOI:10.1002/humu.23701] [PMID]
Perez, D., Hsieh, D. T., & Rohena, L. (2017). Somatic mosaicism of PCDH19 in a male with early infantile epileptic encephalopathy and review of the literature. American Journal of Medical Genetics Part A, 173(6), 1625-1630. [DOI:10.1002/ajmg.a.38233] [PMID]
Rakotomamonjy, J., Sabetfakhri, N. P., McDermott, S. L., & Guemez‐Gamboa, A. (2020). Characterization of seizure susceptibility in Pcdh19 mice. Epilepsia, 61(10), 2313-2320. [DOI:10.1111/epi.16675] [PMID]
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., & Gastier-Foster, J., et al.(2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405-423. [DOI:10.1038/gim.2015.30] [PMID]
Robens, B. K., Yang, X., McGraw, C. M., Turner, L. H., Robens, C., & Thyme, S., et al. (2022). Mosaic and non-mosaic protocadherin 19 mutation leads to neuronal hyperexcitability in zebrafish. Neurobiology of Disease, 169, 105738. [DOI:10.1016/j.nbd.2022.105738] [PMID]
Ryan, S. G., Chance, P. F., Zou, C. H., Spinner, N. B., Golden, J. A., & Smietana, S. (1997). Epilepsy and mental retardation limited to females: An X-linked dominant disorder with male sparing. Nature Genetics, 17(1), 92-95. [DOI:10.1038/ng0997-92] [PMID]
Samanta, D. (2020). PCDH19-related epilepsy syndrome: A comprehensive clinical review. Pediatric Neurology, 105, 3-9. [DOI:10.1016/j.pediatrneurol.2019.10.009] [PMID]
Scheffer, I. E., Turner, S. J., Dibbens, L. M., Bayly, M. A., Friend, K., & Hodgson, B., et al. (2008). Epilepsy and mental retardation limited to females: An under-recognized disorder. Brain, 131(4), 918-927. [DOI:10.1093/brain/awm338] [PMID]
Serratto, G. M., Pizzi, E., Murru, L., Mazzoleni, S., Pelucchi, S., & Marcello, E., et al. (2020). The epilepsy-related protein PCDH19 regulates tonic inhibition, GABAAR kinetics, and the intrinsic excitability of hippocampal neurons. Molecular Neurobiology, 57(12), 5336-5351. [DOI:10.1007/s12035-020-02099-7] [PMID]
Shibata, M., Ishii, A., Goto, A., & Hirose, S. (2021). Comparative characterization of PCDH19 missense and truncating variants in PCDH19-related epilepsy. Journal of Human Genetics, 66(6), 569-578. [DOI:10.1038/s10038-020-00880-z] [PMID]
Steel, D., Symonds, J. D., Zuberi, S. M., & Brunklaus, A. (2017). Dravet syndrome and its mimics: Beyond SCN 1A. Epilepsia, 58(11), 1807-1816. [DOI:10.1111/epi.13889] [PMID]
Stosser, M. B., Lindy, A. S., Butler, E., Retterer, K., Piccirillo-Stosser, C. M., & Richard, G., et al. (2018). High frequency of mosaic pathogenic variants in genes causing epilepsy-related neurodevelopmental disorders. Genetics in Medicine, 20(4), 403-410. [DOI:10.1038/gim.2017.114] [PMID]
Symonds, J. D., & McTague, A. (2020). Epilepsy and developmental disorders: Next generation sequencing in the clinic. European Journal of Paediatric Neurology, 24, 15-23. [DOI:10.1016/j.ejpn.2019.12.008] [PMID]
Syrbe, S. (2022). Developmental and epileptic encephalopathies-therapeutic consequences of genetic testing. Medizinische Genetik, 34(3), 215-224. [DOI:10.1515/medgen-2022-2145] [PMID]
Tan, C., Shard, C., Ranieri, E., Hynes, K., Pham, D. H., & Leach, D., et al. (2015). Mutations of protocadherin 19 in female epilepsy (PCDH19-FE) lead to allopregnanolone deficiency. Human Molecular Genetics, 24(18), 5250-5259. [DOI:10.1093/hmg/ddv245] [PMID]
Terracciano, A., Specchio, N., Darra, F., Sferra, A., Bernardina, B. D., & Vigevano, F., et al. (2012). Somatic mosaicism of PCDH19 mutation in a family with low-penetrance EFMR. Neurogenetics, 13(4), 341-345. [DOI:10.1007/s10048-012-0342-9] [PMID]
Thiffault, I., Farrow, E., Smith, L., Lowry, J., Zellmer, L., & Black, B., et al. (2016). PCDH19‐related epileptic encephalopathy in a male mosaic for a truncating variant. American Journal of Medical Genetics Part A, 170(6), 1585-1589. [DOI:10.1002/ajmg.a.37617] [PMID]
Trivisano, M., Lucchi, C., Rustichelli, C., Terracciano, A., Cusmai, R., & Ubertini, G. M., et al. (2017). Reduced steroidogenesis in patients with PCDH 19‐female limited epilepsy. Epilepsia, 58(6), e91-e95. [DOI:10.1111/epi.13772] [PMID]
Trivisano, M., Pietrafusa, N., Ciommo, V.d, Cappelletti, S., Palma, L.d, & Terracciano, A., et al. (2016). PCDH19-related epilepsy and Dravet Syndrome: Face-off between two early-onset epilepsies with fever sensitivity. Epilepsy Research, 125, 32–36. [DOI:10.1016/j.eplepsyres.2016.05.015] [PMID]
Van Harssel, J. J., Weckhuysen, S., van Kempen, M. J., Hardies, K., Verbeek, N. E., & de Kovel, C. G., et al. (2013). Clinical and genetic aspects of PCDH19-related epilepsy syndromes and the possible role of PCDH19 mutations in males with autism spectrum disorders. Neurogenetics, 14(1), 23-34. [DOI:10.1007/s10048-013-0353-1] [PMID]
Wolverton, T., & Lalande, M. (2001). Identification and characterization of three members of a novel subclass of protocadherins. Genomics, 76(1-3), 66-72. [DOI:10.1006/geno.2001.6592] [PMID]
Xu, X., Yang, X., Wu, Q., Liu, A., Yang, X., & Ye, A. Y., et al. (2015). Amplicon resequencing identified parental mosaicism for approximately 10% of “de novo” SCN1A mutations in children with Dravet syndrome. Human Mutation, 36(9), 861-872. [DOI:10.1002/humu.22819] [PMID]
Type of Study: Original |
Subject:
Cellular and molecular Neuroscience
Received: 2023/04/25 | Accepted: 2023/05/27 | Published: 2024/07/20
Received: 2023/04/25 | Accepted: 2023/05/27 | Published: 2024/07/20
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
