

Volume 14, Issue 5 (September & October 2023)
BCN 2023, 14(5): 701-712 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Thapliyal S, Garg N, Joshi R, Chakrabarti A, Medhi B. Pentylenetetrazole Induced Kindling Model of Refractory Epilepsy: A Proof-of-concept Study to Explore Dose and Time Range of Phenobarbital in Rats. BCN 2023; 14 (5) :701-712
URL: http://bcn.iums.ac.ir/article-1-2373-en.html
URL: http://bcn.iums.ac.ir/article-1-2373-en.html



1- Department of Pharmacology, Postgraduate Institute of Medical Education & Research, Chandigarh, India.
Keywords: Drug resistant epilepsy, Pentylenetetrazole, Phenobarbital, Oxidative stress, Kindling, Hippocampus, Introduction
Full-Text [PDF 1151 kb]
| Abstract (HTML)
Full-Text:
1. Introduction
Epilepsy is a multifaceted brain disorder, characterized by frequent episodes of seizures, which could range from mild muscle jerks to unstoppable convulsions (Kumar et al., 2018). Despite the enormous antiseizure drugs (ASD) available, about 30% of patients experience recurrence of seizures, a condition known as “drug resistance” (Lalitha et al., 2018). According to the International League Against Epilepsy, drug resistance is considered an uncontrollable seizure even after sufficient trials of two tolerated and appropriately chosen ASD schedules (whether as monotherapies or in combination) (Kwan et al., 2010).
Moreover, the mechanisms underlying pharmacoresistance remain obscure due to the lack of adequate experimental models of chronic intractable epilepsy (Löscher, 1997). Therefore, this prompted us to validate a model of drug-resistant epilepsy. The most laborious and widely used animal epilepsy model is kindling, induced via chronic administration of pentylenetetrazole (PTZ) (Singh et al., 2021). It acts as the GABA antagonist (Ergul Erkec & Arihan, 2015) and when given a sub-convulsive dose for a longer period, it causes permanent changes in the brain (Sato et al., 1997). In addition, it leads to alterations in the antioxidant brain’s defense systems which leads to oxidative stress (Ilhan et al., 2004) and neuronal death in the hippocampus (Pavlova et al., 2006).
Two types of protocols are used to evaluate the effects of ASDs on kindling: 1) pre-treatment with drugs, before each PTZ injection and examining their impact on the kindling process (anti-epileptogenic potential) by comparing them with a control group; 2) Drugs are tested on fully kindled rats to determine their anticonvulsant potential (Saha & Chakrabarti, 2014). In the present study, the second method was followed to develop drug- refractoriness in rats because of its potential merits. Firstly, the PTZ kindled rats treated with ASDs model targets the refractory neuropathology of seizure whereas the pre-treatment ASDs before the PTZ kindling model is a consequence of tolerance (ASDs used at sub-therapeutic doses) (Loscher & Schmidt, 2006). Secondly, the kindling time in the latter model (8 weeks vs 9 weeks and 5 days) is shorter as compared to prior kindling models (Singh et al., 2014). One of the essential criteria during the validation of pharmacoresistant models is the maintenance of ASD effective levels (Löscher, 2007). Accordingly, phenobarbital (PB) was used because of its longer half-life and has been reported to maintain its adequate levels in plasma for longer periods (Löscher & Hönack, 1989).
The PB+PTZ model perfectly mimics drug-resistant epilepsy in humans (McNamara et al., 1980). The model suggested that resistance to phenobarbital was initially induced as a result of epileptic seizure and the combination of both receptor desensitization and seizure at a later stage (Jing et al., 2010). Based on these findings, the present study aims to validate a refractory model that targets the epileptogenic process behind the development of drug-resistant epilepsy, which is one of the imperative advantages of this model over other preclinical models. Therefore, we planned to study the effect of 3 different doses of PB (20 mg: below sub-therapeutic; 40 mg: sub-therapeutic dose; and 60 mg: maximum tolerated dose) at 2 different intervals in the refractory model of epilepsy in experimental rats by PTZ-induced kindling.
2. Material and Methods
Chemicals and experimental animals
PTZ was obtained from Sigma, India and PB was received as a gift sample by Harman Finochem Pvt Ltd. Aurangabad for carrying out the study. The study was performed in the Department of Pharmacology, PGIMER, Chandigarh, India. Male Wistar rats were acclimatized for one week before being used in the controlled environment (temperature at 24°C with 12-h light/dark cycles). PTZ was dissolved in saline and administered IP whereas PB was dissolved in carboxymethylcellulose and administered orally.
Study groups
Rats were randomly divided into six groups (Figure 1) as follows: Group 1: saline control group; group 2: vehicle carboxymethylcellulose (CMC) treated group; group 3: PTZ-kindled rats; group 4: PTZ-kindled rats, receiving PB (20 mg/kg); group 5: PTZ-kindled rats receiving PB (40 mg/kg); M 6: PTZ-kindled rats receiving PB (60 mg/kg). An equal number of animals were sacrificed from each group on day 14 and day 28.
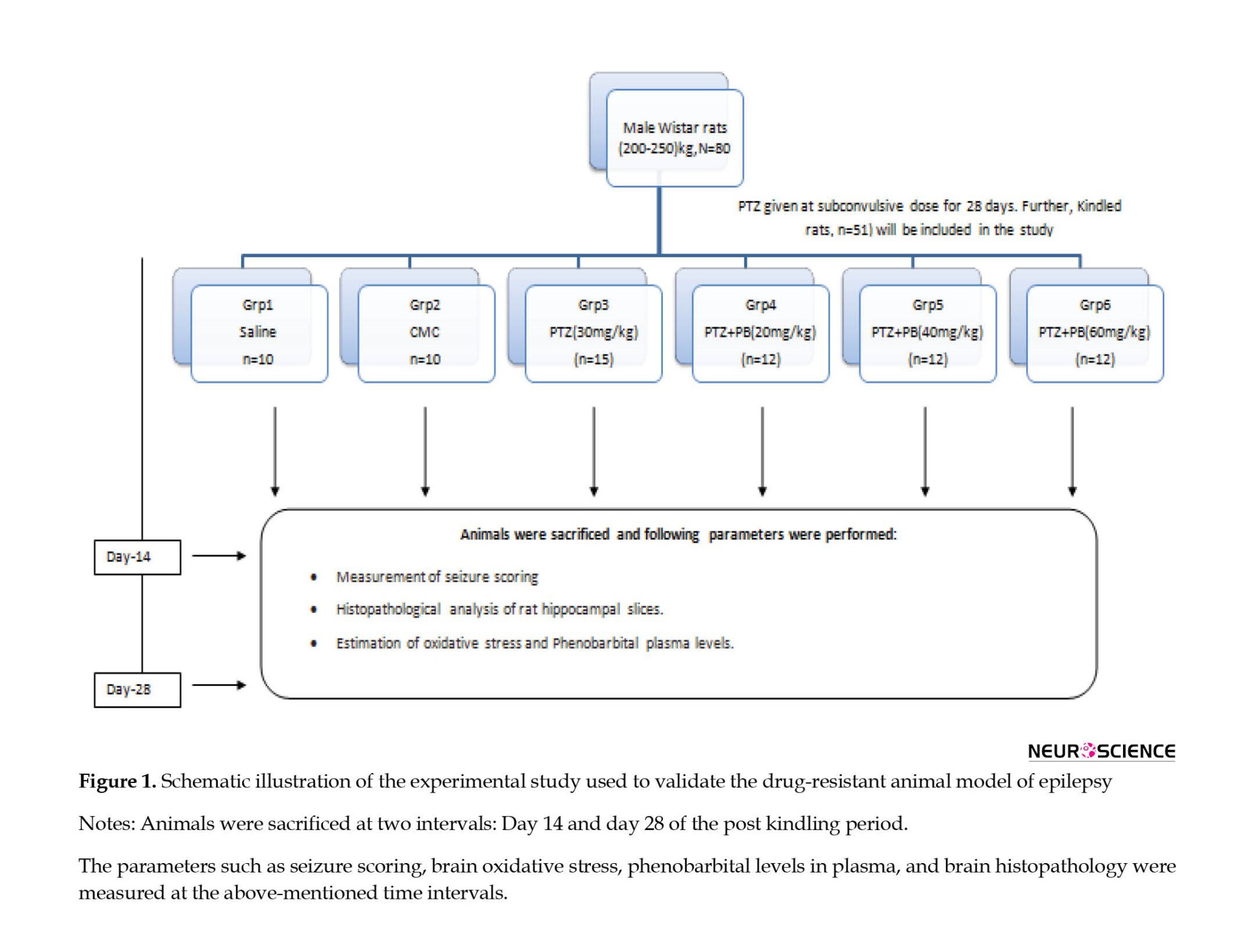
PTZ kindling
The sub-convulsive dose of PTZ (30 mg/kg/day) was given IP for 28 days. The rats were then observed for 30 min for the convulsion activity in a transparent Plexiglass chamber. The seizure scale was followed for seizure scoring (De Sarro et al., 1999, Singh et al., 2017a, Singh et al., 2017b, Singh et al., 2017c, Singh et al., 2016):
0. no response;
1. mouth and facial jerks;
2. nodding or myoclonic body jerks; 3-forelimb clonus;
4. rearing, falling, hind limb clonus and forelimb tonus
5. a tonic extension of the hind limb, status epileptic, and/or death observed for 30 min.
Biochemical estimation
The degree of brain oxidative stress was determined using free carbonyl protein groups as biomarkers, and its measurement was performed according to the spectrophotometric assay. The levels of reduced glutathione, catalase, and superoxide dismutase (SOD) were estimated in rat brain homogenate.
Estimation of reduced glutathione levels
The Elman method (Ellman, 1959) was followed to estimate the levels of reduced glutathione. The proteins were precipitated by adding 10% trichloroacetic acid to an equal quantity of homogenate. The homogenate was then centrifuged for 10 min at 2000 rpm. For 100 µL of protein-free supernatant, firstly 0.3 M phosphate buffer, pH 8.4 (2 mL) was added then 0.04% DTNB was prepared in 1% tri-sodium citrate, and 0.5 mL was added. Thirdly, distilled water (0.4 mL) was added to the supernatant. The glutathione concentration was determined by running a parallel standard GSH. The concentration of reduced glutathione as µg/g-wet tissue was observed spectrophotometrically at 412 nm within 15 min.
Estimation of superoxide dismutase levels/activity
The standard method (Kono. 1978) was followed to estimate SOD levels. The supernatant (0.1 mL) was taken and the ice-cold chloroform (0.25 mL) and methanol (0.15 mL) were added. The mixture was centrifuged at 3000 rpm for 10 min at 4°C. The supernatant was separated and 0.2 mL of the supernatant, 0.5 mL ethylene diamine tetraacetic acid, 1.3 mL buffer, and 0.8 mL water was added respectively in order. The addition of epinephrine (0.2 mL) started the reaction. The absorbance was read at every 30 s for 3 min at 480 nm and then the average change in absorbance was calculated. The data was expressed as nmol/min/mg protein.
Estimation/determination of catalase activity
The catalase activity of the brain tissue was measured as described by (Greenwald, 2017). Briefly, 0.05 mL of brain homogenate was used for the reaction mixture. In the homogenate, 0.05 M phosphate buffer, pH 7.0 (1.95 mL), and 0.019 M hydrogen peroxide (1.0 mL) were added for the final volume to make up the 3 mL. The absorbance of the reaction mixture was calculated at 240 nm after a 60-s interval for 2 min. The catalase activity was expressed in μmol/mg protein.
Estimation of plasma levels of phenobarbital
The levels of phenobarbital were estimated via the high-performance liquid chromatography analysis. Animals were sacrificed at two different time points, namely day 14 and day 28 after kindling, and blood was collected retroorbitally. The elution of phenobarbital was done from a reversed-phase column with an optimum temperature of 50°C. The mobile phase used consisted of 19/81 by the vol acetonitrile/phosphate buffer and the flow rate was set at 3.0 mL/min. The absorption of phenobarbital occurred at 195 nm and its quantity was estimated from the respective peak height. The sample injection volume was 10 μL. The time required for each analysis was about 14 min (Moriyama, 1999).
Histopathology of rat brain
The brain tissues were stored in 10% neutral formalin for the histopathological analysis. The brain tissues were processed by treating them with various concentrations of alcohol to remove the water from the tissues. The brain sections were then embedded in a molten wax and thin sections were cut (5 μm) and fixed on a glass slide. Then, it was stained with hematoxylin and eosin dye. Bright-field images were taken using an Olympus upright microscope.
The neuronal injury score was determined according to the following criteria: normal=no injury or rare isolated apoptotic neuron was given the score of 0; Rare neuronal injury less than 5 clusters=Score 1; The occasional neuronal injury (5-15 clusters)=Score 2; The frequent neuronal injury (>15 clusters)=Score 3; Diffuse neuronal injury=Score 4 (Wyler, 1992).
Statistical analysis
All data were expressed as Mean±SD. The quantitative parameters, such as seizure score and oxidative stress parameters were evaluated using a one-way analysis of variance (ANOVA) and the Bonferroni post hoc analysis. The unpaired student t-test was performed to compare the plasma concentration of different doses of PB. P<0.05 was considered statistically significant.
3. Results
Seizure scoring in PTZ kindled rats after the administration of different doses of phenobarbital for different periods
The rats in the saline as well as the CMC-treated groups demonstrated 0 seizure score throughout the 4-week kindling period. In the PTZ-treated group (30 mg/kg), the seizure score increased from 2.67±0.44 to 3.3±0.57 during the study period (Figure 2). Kindled rats treated with PB (20 mg/kg) showed no significant decline in the seizure score at the end of day 14 as well as day 28 of the study period. The phenobarbital 40 mg/kg (P<0.05) and 60 mg/kg (P<0.01) showed a significant reduction in the seizure score at the end of day 14 in the PTZ kindled animals. However, the chronic treatment of PB at the dose of 40 mg/kg and 60 mg/kg along with PTZ administration produced resistance as the seizure scores were found to be increased at the end of day 28 (P<0.05) (Figure 2).

Effects of phenobarbital treatment on reduced glutathione levels
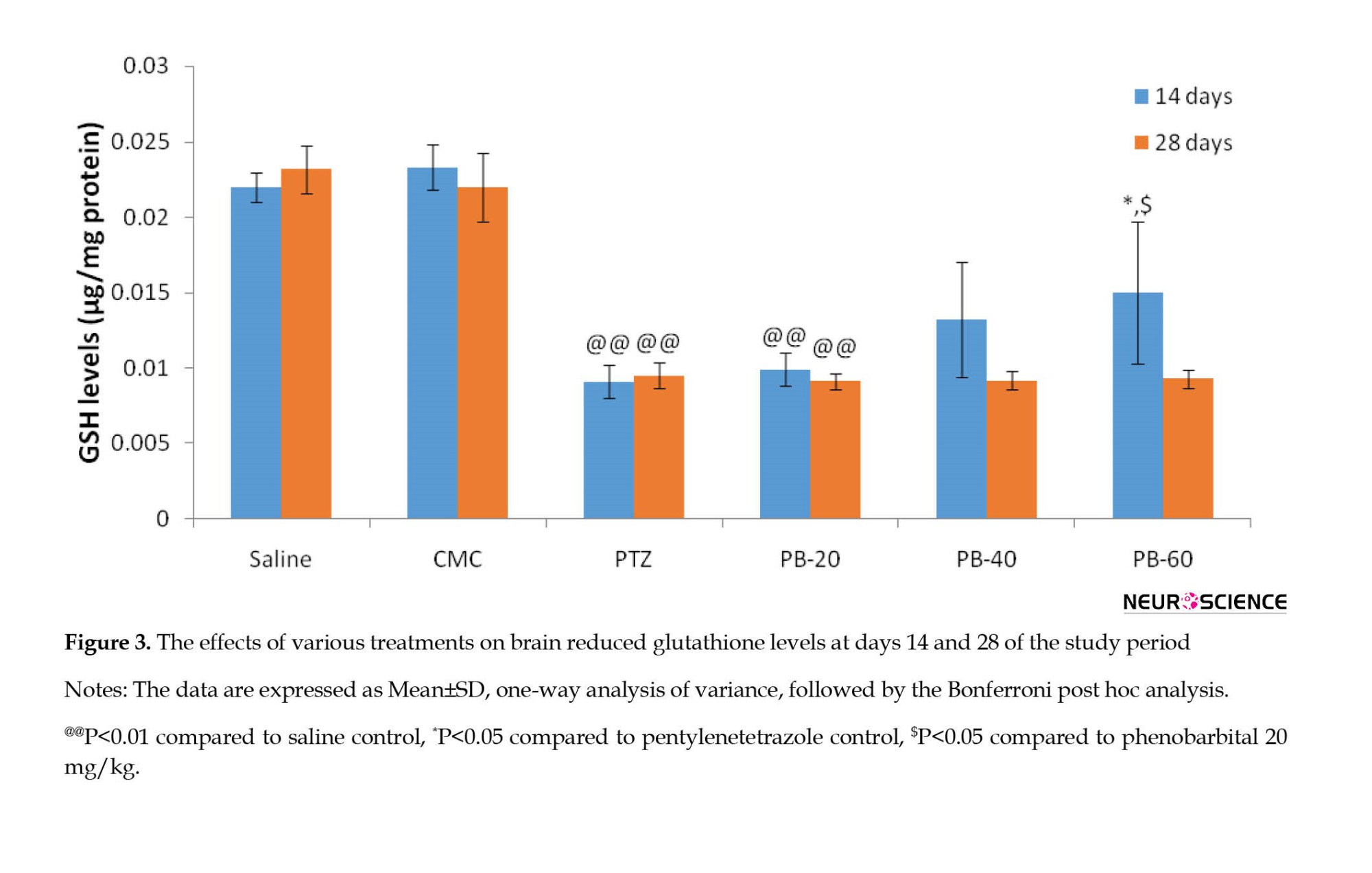
The glutathione levels in rat brains were remarkably higher (P<0.05) in the PB 60 mg/kg treated group compared to the PTZ treated group and no significant effects were observed at PB dose 20 mg/kg and 40 mg/kg at the end of day 14. At the end of day 28 of PB treatment, no significant effects were observed in glutathione levels in all three treatment groups as compared to PTZ treated group (Figure 3).
Effects of phenobarbital treatment on superoxide dismutase levels
The level of SOD in rat brain was significantly reduced (P<0.05) in the PTZ treatment group as compared to vehicle, saline, and treated groups at the end of both 14 and 28 days of PB treatment. The decrease in the SOD levels was alleviated (P<0.05) with the treatment of PB (40 and 60 mg/kg) at the end of day 14. However, the chronic administrations of PB along with PTZ for 28 days in the kindled rats lead to a reduction in the levels of SOD (Figure 4).

Effect of phenobarbital treatment on catalase
The catalase levels in rat brains were significantly decreased (P<0.05) in PTZ treated group as compared to both the vehicle and saline-treated groups. The catalase levels were significantly increased (P<0.05) with PB treatment (40 and 60 mg/kg) dose on day 14. The treatment of PB (40 and 60 mg/kg) reduced the brain catalase levels as compared to the normal control group at the end of day 28 of PB treatment. However, this decrease in CAT level was not statistically significant (Figure 5). The brain catalase levels of phenobarbital 20 mg/kg group showed no significant effects when compared to PTZ control at day 28 of PB+PTZ treatment.

Effect on plasma levels of phenobarbital
The concentration of different doses of PB (20, 40, and 60 mg/kg) was measured in the rat’s blood at the end of day 14 day 28 of treatment with PB. The PB treatment at the dose of 20 mg/kg showed no significant increment in its plasma at the end of 28 days. However, a highly significant increase (P<0.01) in the PB levels was observed with the treatment of 40 mg/kg at the end of day 28 as compared to day 14 of PB treatment in PTZ kindled rats. The plasma levels of PB were significantly increased after 28 days of treatment at the dose of 60 mg/kg as compared to PTZ kindled rats. Moreover, the plasma concentration of PB was remarkably higher (P<0.05) in the 60 mg/kg PB treatment group in contrast to the 20 mg/kg PB treatment group at the end of day 28 (Table 1).

Neuronal injury score
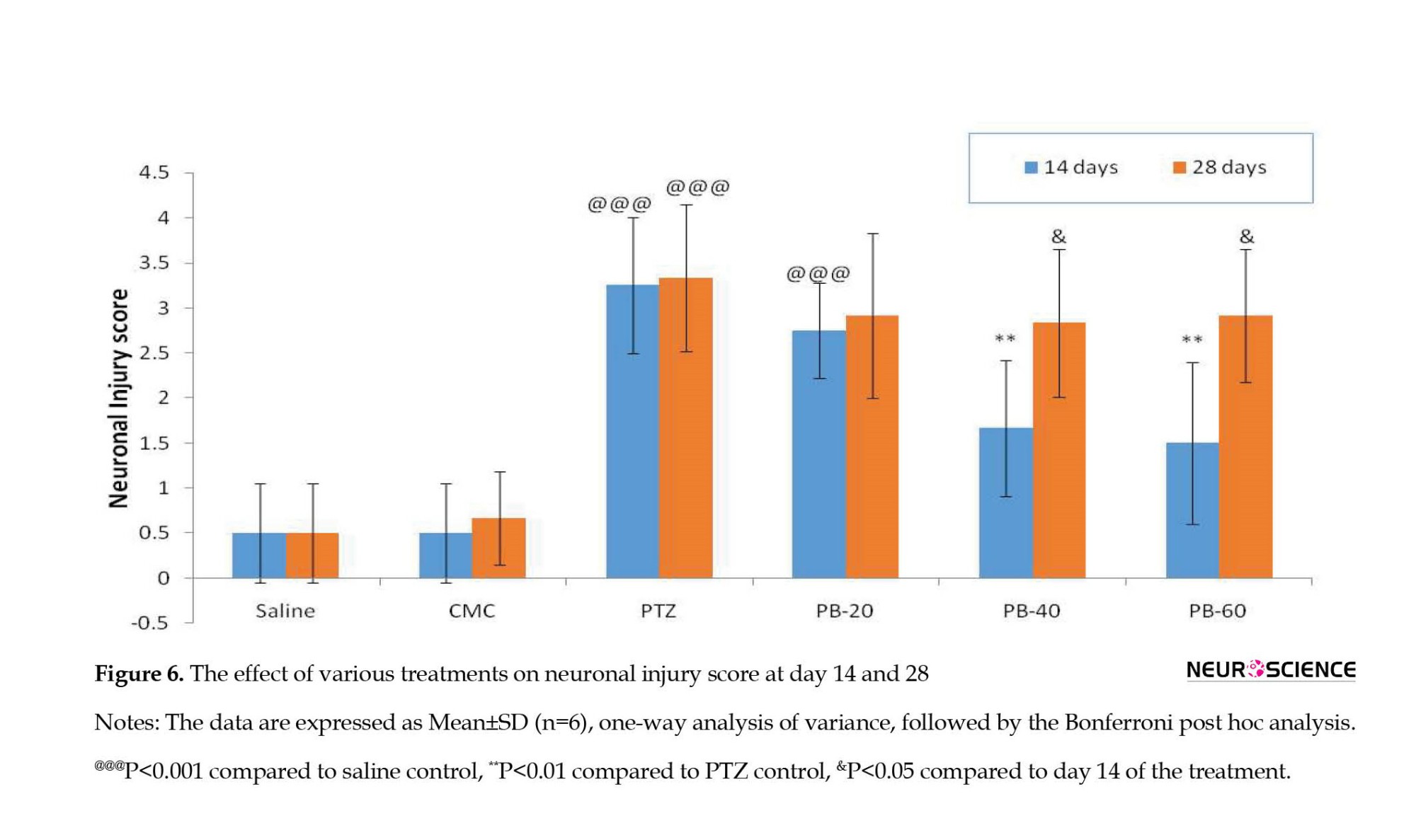
The PTZ-treated group showed a significantly higher (P<0.001) neuronal injury score in comparison to the normal saline control group. The treatment of PB (40 and 60 mg/kg) significantly reduced (P<0.01) the neuronal injury score as compared to the PTZ-treated group at the end of day 14. However, the combination of PTZ and chronic administration of phenobarbital for 28 days increased the seizure severity and neuronal injury score leading to PB resistance. Thus, it was observed that the treatment of PB (40 and 60 mg/kg) reversed the decrease (P<0.05) in neuronal injury score at the end of day 28 when compared to day 14 of the PB+PTZ treatment period (Figure 6).
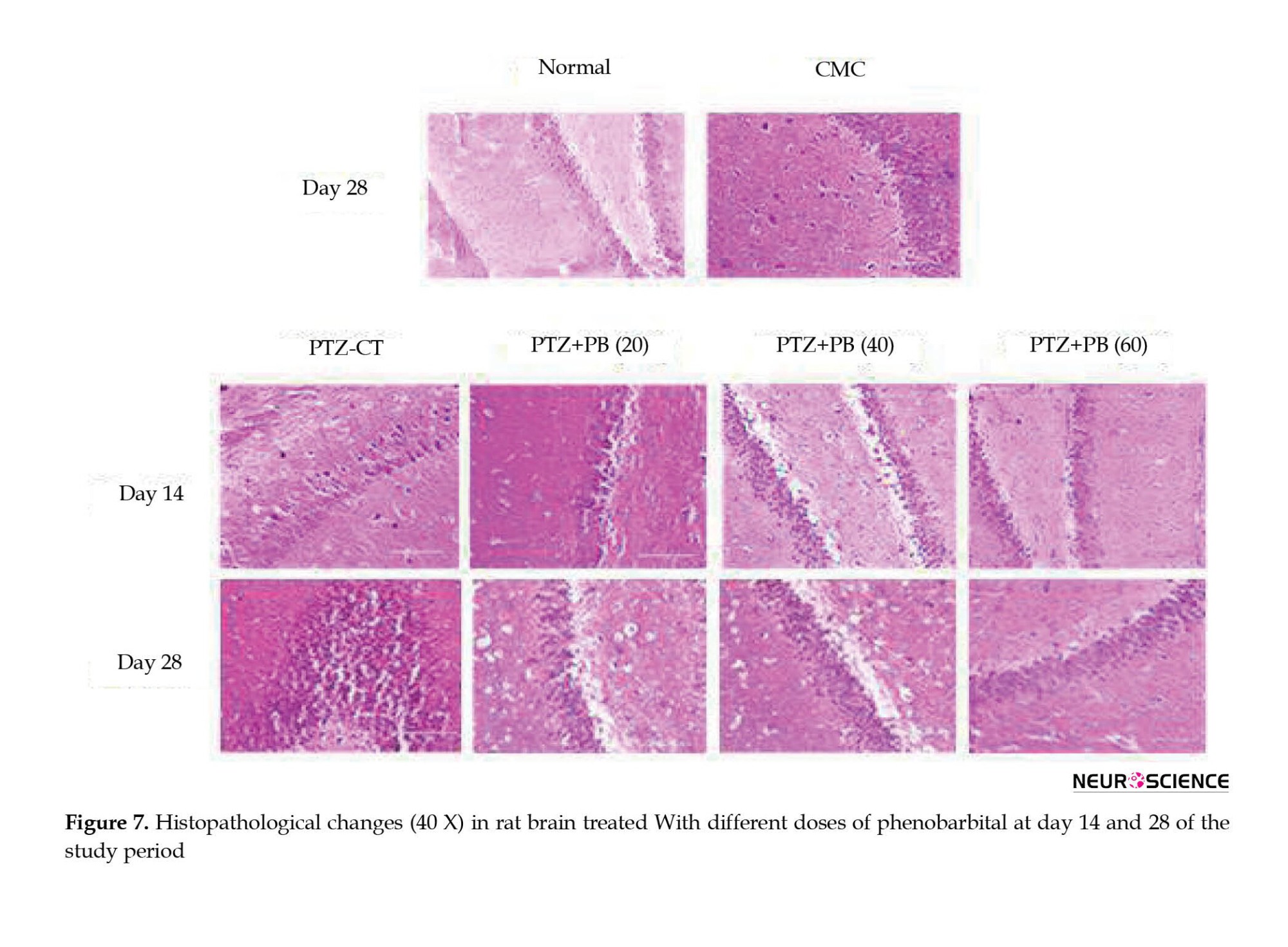
The PTZ-treated group showed a diffuse neuronal injury as compared to the normal saline group showing normal neurons with rare neuronal injury (Figure 7). The treatment of PB at 20 mg/kg dose showed frequent neuronal injury with (>15 clusters) at the end of 14 as well as 28 days of the study period. The occasional neuronal injury with (5-15 clusters) of neurons was observed with treatment of PB (40 and 60 mg/kg) at the end of day 14 of the study period. However, at the end of day 28, frequent neuronal injury with (>15 clusters) was observed with 40 and 60 mg/kg doses of PB treatment.
4. Discussion
This is the first study to elucidate and report the different parameters studied on PTZ-kindled Wistar rats at two different time points with three different doses of PB to select the dose at which maximum resistance occurs within a chronic period. The current research work reveals that phenobarbital treatment at 20 mg/kg dose showed no protective effect on seizure severity at any time point. The PB administration at both 40 mg/kg and 60 mg/kg doses showed an initial decrease in the seizure score and later developed resistance with an increase in seizure severity. This might be due to an increase in P-glycoprotein which is triggered by both seizure and the drug effect, thus leading to refractoriness (Jing et al., 2010). Moreover, drug tolerance is also one of the phenomena which leads to resistance. For example, phenobarbital undergoes pharmacokinetic tolerance which is attributed to its fast metabolism rate (Gay et al., 1983). Hence, in the current paper dose escalation (20 mg, 40 mg, 60 mg) method was used to combat tolerance at two (day 14 and day 28) time intervals. Increased dose helps in maintaining the same antiepileptic efficacy as observed earlier with low-dose treatment. The above-described tolerance is detected through plasma-level monitoring (Loscher & Schmidt, 2006). Following previous studies, a similar increase in the trend of PB levels in plasma has been observed at day 14 and day 28 in our research (Jing et al., 2010). However, one preclinical study on the spontaneous recurrent seizures model of drug-resistant epilepsy has also reported no differentiable comparison between plasma levels of PB responders and non-responders’ rodents (Brandt et al., 2004). PB epileptic brain is more prone to adverse effects because of the permanent alteration imprinted in the brain during epileptogenesis. There are studies reporting symptoms of sleepiness, dizziness, altered behavior, and depression as associated side effects among people with epilepsy taking PB at higher doses (Zhang et al., 2011). Therefore, to avoid the associated side effects 40 mg/kg was advised over 60 mg/kg, as the perfect dose of PB to induce resistance model which can easily be extrapolated in the clinical scenario.
The present pharmaco-resistant animal model has an advantage over conventional amygdala kindling and spontaneous recurrent seizures followed by PB and phenytoin treatment. It is less time-consuming and labor-intensive and could screen compounds over a short period. Moreover, no surgical expertise and electrode implants are required in the current animal model, which further decreases the chances of animal mortality. The major difference between our model and another promising pharmaco-resistance model is different in the study period. Our research model was a 56-day chronic study which is better suited to studying drug-resistant mechanisms. Additionally, chronic brain alteration can change the pharmacological response that cannot be visualized in acute models (Löscher, 2017).
Oxidative stress due to free radicals plays a pivotal role in the neurobiology of epilepsy. Recurrent seizures lead to the activation of the oxidative defense system and the production of free radicals (Shin et al., 2011). Further, the activation of free radicals leads to alteration in the levels of endogenous antioxidant enzymes, lipids, and expression of DNA (Ergul Erkec & Arihan, 2015). Numerous studies have depicted the involvement of certain endogenous (glutathione, catalase, superoxide dismutase) enzymes with neurodegenerative disorders (Cardenas-Rodriguez, 2013). Previous evidence supports the relationship between oxidative stress and mechanisms that lead to drug–resistance (Lorigados Pedre et al., 2018). The present study showed that the levels of antioxidant enzymes (glutathione, catalase, superoxide dismutase) in the PB 40 and 60 mg/kg treatment groups were decreased at the end of 28 days indicating the increase in oxidative stress, which depicts its positive correlation with drug-resistance epilepsy.
Previous reports have suggested that the majority of PB-resistant rats lead to major hippocampal damage (Bethmann, 2007). Based on preclinical findings it has been proposed that patients with uncontrollable seizures will have severe hippocampal damage as compared to patients with controlled epilepsy (Sloviter, 1994). Such clinical studies have found progressive damage to the hippocampus of patients with drug-resistant epilepsy (Kalviainen, 1998). Similar findings have been observed in our study where the combination of PTZ and chronic administration of the PB group has shown an increase in the neuronal injury score at the end of 28 days contributing to the process of epileptogenesis and the development of the model of drug resistance.
5. Conclusion
Drug resistance is the most challenging problem in the management of epilepsy. Therefore, it is necessary to understand the multifactorial mechanisms of epileptogenesis and develop animal models that target epileptogenesis for the better management of refractory epilepsy. Further, PTZ+PB (40 mg/kg) has shown the best results and it could be used to develop the refractory model of epilepsy and also to screen and study the various combinations of drugs that would help prevent and treat the resistance epilepsy.
Ethical Considerations
Compliance with ethical guidelines
All the experiments were performed according to the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), India. The study was approved by the Institutional Animal Ethics Committee and Institutional Biosafety Committee (No.: JSSCP/IAEC/OT/M.PHARM/PH.COLOGY/04/17-18).
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
Conceptualization: Bikash Medhi; Methodology: Nitika Garg, Amitava Chakrabarti, Bikash Medhi; Formal analysis and investigation: Surabhi Thapliyal, Nitika Garg; Writing- original draft preparation: Surabhi Thapliyal, Nitika Garg and Rupa Joshi; Review and editing: Nitika Garg, Rupa Joshi, Amitava Chakrabarti and Bikash Medhi; Supervision: Amitava Chakrabarti and Bikash Medhi.
Conflict of interest
The authors declared no conflict of interest.
Acknowledgments
The authors acknowledge the support from Experimental Pharmacology Laboratory (EPL) and PGIMER Chandīgarh, for providing all the experimental facilities to conduct the research.
References
Bethmann, K., Brandt, C., & Löscher, W. (2007). Resistance to phenobarbital extends to phenytoin in a rat model of temporal lobe epilepsy. Epilepsia, 48(4), 816-826. [DOI:10.1111/j.1528-1167.2007.00980.x] [PMID]
Brandt, C., Volk, H. A., & Löscher, W. (2004). Striking differences in individual anticonvulsant response to phenobarbital in rats with spontaneous seizures after status epilepticus. Epilepsia, 45(12), 1488–1497. [DOI:10.1111/j.0013-9580.2004.16904.x] [PMID]
Cardenas-Rodriguez, N., Huerta-Gertrudis, B., Rivera-Espinosa, L., Montesinos-Correa, H., Bandala, C., & Carmona-Aparicio, L., et al (2013). Role of Oxidative stress in refractory epilepsy: Evidence in patients and experimental models. International Journal of Molecular Sciences, 14(1), 1455–1476. [DOI:10.3390/ijms14011455] [PMID]
De Sarro, A., Naccari, F., & De Sarro, G. (1999). Enhanced susceptibility of pentylenetetrazole kindled mice to quinolone effects. International Journal of Antimicrobial Agents, 12(3), 239–244. [DOI:10.1016/S0924-8579(99)00067-9] [PMID]
Ellman, G. L. (1959). Tissue sulfhydryl groups. Archives of Biochemistry and Biophysics, 82(1), 70–77. [DOI:10.1016/0003-9861(59)90090-6] [PMID]
Ergul Erkec, O., & Arihan, O. (2015). Pentylenetetrazol kindling epilepsy model. Turkish Epilepsy Society, 21(1), 6-12 [DOI:10.5505/epilepsi.2015.08108]
Gay, M. H., Ryan, G. P., Boisse, N. R., & Guarino, J. J. (1983). Phenobarbital tolerance and physical dependence: Chronically equivalent dosing model. European Journal of Pharmacology, 95(1-2), 21–29. [DOI:10.1016/0014-2999(83)90263-7] [PMID]
Greenwald, R. A. (2017). Handbook methods for oxygen radical research. Boca Raton: CRC Press. [Link]
Ilhan, A., Iraz, M., Gurel, A., Armutcu, F., & Akyol, O. (2004). Caffeic acid phenethyl ester exerts a neuroprotective effect on CNS against pentylenetetrazol-induced seizures in mice. Neurochemical Research, 29(12), 2287–2292. [DOI:10.1007/s11064-004-7038-y] [PMID]
Jing, X., Liu, X., Wen, T., Xie, S., Yao, D., & Liu, X., et al (2010). Combined effects of epileptic seizure and phenobarbital induced overexpression of P-glycoprotein in brain of chemically kindled rats. British Journal of Pharmacology, 159(7), 1511–1522. [DOI:10.1111/j.1476-5381.2009.00634.x] [PMID]
Kälviäinen, R., Salmenperä, T., Partanen, K., Vainio, P., Riekkinen, P., & Pitkänen, A. (1998). Recurrent seizures may cause hippocambal damage in temporal lobe epilepsy. Neurology, 50(5), 1377–1382. [DOI:10.1212/WNL.50.5.1377] [PMID]
Kono, Y. (1978). Generation of superoxide radical during autoxidation of hydroxylamine and an assay for superoxide dismutase. Archives of Biochemistry and Biophysics, 186(1), 189–195. [DOI:10.1016/0003-9861(78)90479-4] [PMID]
Kumar, B., Medhi, B., Modi, M., Saikia, B., Attri, S. V., & Patial, A. (2018). A mechanistic approach to explore the neuroprotective potential of zonisamide in seizures. Inflammopharmacology, 26(4), 1125–1131. [DOI:10.1007/s10787-018-0478-9] [PMID]
Kwan, P., Arzimanoglou, A., Berg, A. T., Brodie, M. J., Allen Hauser, W., & Mathern, G., et al. (2010). Definition of drug resistant epilepsy: Consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia, 51(6), 1069–1077. [DOI:10.1111/j.1528-1167.2009.02397.x] [PMID]
Lalitha, S., Minz, R. W., & Medhi, B. (2018). Understanding the controversial drug targets in epilepsy and pharmacoresistant epilepsy. Reviews in The Neurosciences, 29(3), 333–345. [DOI:10.1515/revneuro-2017-0043] [PMID]
Lorigados Pedre, L., Gallardo, J. M., Morales Chacón, L. M., Vega García, A., Flores-Mendoza, M., & Neri-Gómez, T., et al. (2018). Oxidative stress in patients with drug resistant partial complex seizure. Behavioral Sciences (Basel, Switzerland), 8(6), 59. [PMID]
Löscher, W. (2017). Animal models of drug-refractory epilepsy. In: A. Pitkänen, P. S. Buckmaster, & S. L. Moshé (Es.), Models of seizures and epilepsy (pp. 43-60). Massachusetts: Academic Press [DOI:10.1016/B978-0-12-804066-9.00051-1]
Löscher, W. (2007). The pharmacokinetics of antiepileptic drugs in rats: Consequences for maintaining effective drug levels during prolonged drug administration in rat models of epilepsy. Epilepsia, 48(7), 1245–1258. [DOI:10.1111/j.1528-1167.2007.01093.x] [PMID]
Löscher, W., & Schmidt, D. (2006). Experimental and clinical evidence for loss of effect (tolerance) during prolonged treatment with antiepileptic drugs. Epilepsia, 47(8), 1253-1284. [DOI:10.1111/j.1528-1167.2006.00607.x] [PMID]
Löscher, W. (1997). Animal models of intractable epilepsy. Progress in Neurobiology, 53(2), 239–258. [DOI:10.1016/S0301-0082(97)00035-X] [PMID]
Löscher, W., & Hönack, D. (1989). Comparison of the anticonvulsant efficacy of primidone and phenobarbital during chronic treatment of amygdala-kindled rats. European Journal of Pharmacology, 162(2), 309–322. [DOI:10.1016/0014-2999(89)90294-X] [PMID]
McNamara, J. O., Byrne, M. C., Dasheiff, R. M., & Fitz, J. G. (1980). The kindling model of epilepsy: A review. Progress in Neurobiology, 15(2), 139–159. [DOI:10.1016/0301-0082(80)90006-4] [PMID]
Moriyama, M., Yamashita, S., Domoto, H., Furuno, K., Araki, H., & Gomita, Y. (1999). Determination of plasma phenobarbital concentration by high-performance liquid chromatography in rat offspring. Journal of Chromatography. B, Biomedical Sciences and Applications, 723(1-2), 301–305. [DOI:10.1016/S0378-4347(98)00534-9] [PMID]
Pavlova, T., Stepanichev, M., & Gulyaeva, N. (2006). Pentylenetetrazole kindling induces neuronal cyclin B1 expression in rat hippocampus. Neuroscience Letters, 392(1-2), 154–158.[DOI:10.1016/j.neulet.2005.09.021] [PMID]
Saha, L., & Chakrabarti, A. (2014). Understanding the anti-kindling role and its mechanism of Resveratrol in Pentylenetetrazole induced-kindling in a rat model. Pharmacology, Biochemistry, and Behavior, 120, 57–64. [DOI:10.1016/j.pbb.2014.01.010] [PMID]
Sato, M., Racine, R. J., & McIntyre, D. C. (1990). Kindling: Basic mechanisms and clinical validity. Electroencephalography and Clinical Neurophysiology, 76(5), 459-472. [DOI:10.1016/0013-4694(90)90099-6]
Shin, E. J., Jeong, J. H., Chung, Y. H., Kim, W. K., Ko, K. H., & Bach, J. H., et al. (2011). Role of oxidative stress in epileptic seizures. Neurochemistry International, 59(2), 122–137. [DOI:10.1016/j.neuint.2011.03.025] [PMID]
Singh, E., Pillai, K. K., & Mehndiratta, M. (2014). Characterization of a lamotrigine-resistant kindled model of epilepsy in mice: Evaluation of drug resistance mechanisms. Basic & Clinical Pharmacology & Toxicology, 115(5), 373–378. [DOI:10.1111/bcpt.12238] [PMID]
Singh, T., Mishra, A., & Goel, R. K. (2021). PTZ kindling model for epileptogenesis, refractory epilepsy, and associated comorbidities: Relevance and reliability. Metabolic Brain Disease, 36(7), 1573-1590. [DOI:10.1007/s11011-021-00823-3] [PMID]
Singh, T., Kaur, T., & Goel, R. K. (2017). Ferulic acid supplementation for management of depression in epilepsy. Neurochemical Research, 42(10), 2940–2948. [DOI:10.1007/s11064-017-2325-6] [PMID]
Singh, T., Kaur, T., & Goel, R. K. (2017). Adjuvant quercetin therapy for combined treatment of epilepsy and comorbid depression. Neurochemistry International, 104, 27–33. [DOI:10.1016/j.neuint.2016.12.023] [PMID]
Singh, T., & Goel, R. K. (2017). Adjuvant neuronal nitric oxide synthase inhibition for combined treatment of epilepsy and comorbid depression. Pharmacological Reports: PR, 69(1), 143–149. [DOI:10.1016/j.pharep.2016.10.001] [PMID]
Singh, T., & Goel, R. K. (2016). Adjuvant indoleamine 2,3-dioxygenase enzyme inhibition for comprehensive management of epilepsy and comorbid depression. European Journal of Pharmacology, 784, 111–120. [DOI:10.1016/j.ejphar.2016.05.019] [PMID]
Sloviter R. S. (1994). The functional organization of the hippocampal dentate gyrus and its relevance to the pathogenesis of temporal lobe epilepsy. Annals of Neurology, 35(6), 640–654.[DOI:10.1002/ana.410350604] [PMID]
Wyler, A. R., Curtis Dohan, F., Schweitzer, J. B., & Berry, A. D. (1992). A grading system for mesial temporal pathology (hippocampal sclerosis) from anterior temporal lobectomy. Journal of Epilepsy, 5(4), 220-225. [DOI:10.1016/S0896-6974(05)80120-3]
Zhang, L. L., Zeng, L. N., & Li, Y. P. (2011). Side effects of phenobarbital in epilepsy: A systematic review. Epileptic Disorders: International Epilepsy Journal with Videotape, 13(4), 349–365. [DOI:10.1684/epd.2011.0444] [PMID]
Epilepsy is a multifaceted brain disorder, characterized by frequent episodes of seizures, which could range from mild muscle jerks to unstoppable convulsions (Kumar et al., 2018). Despite the enormous antiseizure drugs (ASD) available, about 30% of patients experience recurrence of seizures, a condition known as “drug resistance” (Lalitha et al., 2018). According to the International League Against Epilepsy, drug resistance is considered an uncontrollable seizure even after sufficient trials of two tolerated and appropriately chosen ASD schedules (whether as monotherapies or in combination) (Kwan et al., 2010).
Moreover, the mechanisms underlying pharmacoresistance remain obscure due to the lack of adequate experimental models of chronic intractable epilepsy (Löscher, 1997). Therefore, this prompted us to validate a model of drug-resistant epilepsy. The most laborious and widely used animal epilepsy model is kindling, induced via chronic administration of pentylenetetrazole (PTZ) (Singh et al., 2021). It acts as the GABA antagonist (Ergul Erkec & Arihan, 2015) and when given a sub-convulsive dose for a longer period, it causes permanent changes in the brain (Sato et al., 1997). In addition, it leads to alterations in the antioxidant brain’s defense systems which leads to oxidative stress (Ilhan et al., 2004) and neuronal death in the hippocampus (Pavlova et al., 2006).
Two types of protocols are used to evaluate the effects of ASDs on kindling: 1) pre-treatment with drugs, before each PTZ injection and examining their impact on the kindling process (anti-epileptogenic potential) by comparing them with a control group; 2) Drugs are tested on fully kindled rats to determine their anticonvulsant potential (Saha & Chakrabarti, 2014). In the present study, the second method was followed to develop drug- refractoriness in rats because of its potential merits. Firstly, the PTZ kindled rats treated with ASDs model targets the refractory neuropathology of seizure whereas the pre-treatment ASDs before the PTZ kindling model is a consequence of tolerance (ASDs used at sub-therapeutic doses) (Loscher & Schmidt, 2006). Secondly, the kindling time in the latter model (8 weeks vs 9 weeks and 5 days) is shorter as compared to prior kindling models (Singh et al., 2014). One of the essential criteria during the validation of pharmacoresistant models is the maintenance of ASD effective levels (Löscher, 2007). Accordingly, phenobarbital (PB) was used because of its longer half-life and has been reported to maintain its adequate levels in plasma for longer periods (Löscher & Hönack, 1989).
The PB+PTZ model perfectly mimics drug-resistant epilepsy in humans (McNamara et al., 1980). The model suggested that resistance to phenobarbital was initially induced as a result of epileptic seizure and the combination of both receptor desensitization and seizure at a later stage (Jing et al., 2010). Based on these findings, the present study aims to validate a refractory model that targets the epileptogenic process behind the development of drug-resistant epilepsy, which is one of the imperative advantages of this model over other preclinical models. Therefore, we planned to study the effect of 3 different doses of PB (20 mg: below sub-therapeutic; 40 mg: sub-therapeutic dose; and 60 mg: maximum tolerated dose) at 2 different intervals in the refractory model of epilepsy in experimental rats by PTZ-induced kindling.
2. Material and Methods
Chemicals and experimental animals
PTZ was obtained from Sigma, India and PB was received as a gift sample by Harman Finochem Pvt Ltd. Aurangabad for carrying out the study. The study was performed in the Department of Pharmacology, PGIMER, Chandigarh, India. Male Wistar rats were acclimatized for one week before being used in the controlled environment (temperature at 24°C with 12-h light/dark cycles). PTZ was dissolved in saline and administered IP whereas PB was dissolved in carboxymethylcellulose and administered orally.
Study groups
Rats were randomly divided into six groups (Figure 1) as follows: Group 1: saline control group; group 2: vehicle carboxymethylcellulose (CMC) treated group; group 3: PTZ-kindled rats; group 4: PTZ-kindled rats, receiving PB (20 mg/kg); group 5: PTZ-kindled rats receiving PB (40 mg/kg); M 6: PTZ-kindled rats receiving PB (60 mg/kg). An equal number of animals were sacrificed from each group on day 14 and day 28.
PTZ kindling
The sub-convulsive dose of PTZ (30 mg/kg/day) was given IP for 28 days. The rats were then observed for 30 min for the convulsion activity in a transparent Plexiglass chamber. The seizure scale was followed for seizure scoring (De Sarro et al., 1999, Singh et al., 2017a, Singh et al., 2017b, Singh et al., 2017c, Singh et al., 2016):
0. no response;
1. mouth and facial jerks;
2. nodding or myoclonic body jerks; 3-forelimb clonus;
4. rearing, falling, hind limb clonus and forelimb tonus
5. a tonic extension of the hind limb, status epileptic, and/or death observed for 30 min.
Biochemical estimation
The degree of brain oxidative stress was determined using free carbonyl protein groups as biomarkers, and its measurement was performed according to the spectrophotometric assay. The levels of reduced glutathione, catalase, and superoxide dismutase (SOD) were estimated in rat brain homogenate.
Estimation of reduced glutathione levels
The Elman method (Ellman, 1959) was followed to estimate the levels of reduced glutathione. The proteins were precipitated by adding 10% trichloroacetic acid to an equal quantity of homogenate. The homogenate was then centrifuged for 10 min at 2000 rpm. For 100 µL of protein-free supernatant, firstly 0.3 M phosphate buffer, pH 8.4 (2 mL) was added then 0.04% DTNB was prepared in 1% tri-sodium citrate, and 0.5 mL was added. Thirdly, distilled water (0.4 mL) was added to the supernatant. The glutathione concentration was determined by running a parallel standard GSH. The concentration of reduced glutathione as µg/g-wet tissue was observed spectrophotometrically at 412 nm within 15 min.
Estimation of superoxide dismutase levels/activity
The standard method (Kono. 1978) was followed to estimate SOD levels. The supernatant (0.1 mL) was taken and the ice-cold chloroform (0.25 mL) and methanol (0.15 mL) were added. The mixture was centrifuged at 3000 rpm for 10 min at 4°C. The supernatant was separated and 0.2 mL of the supernatant, 0.5 mL ethylene diamine tetraacetic acid, 1.3 mL buffer, and 0.8 mL water was added respectively in order. The addition of epinephrine (0.2 mL) started the reaction. The absorbance was read at every 30 s for 3 min at 480 nm and then the average change in absorbance was calculated. The data was expressed as nmol/min/mg protein.
Estimation/determination of catalase activity
The catalase activity of the brain tissue was measured as described by (Greenwald, 2017). Briefly, 0.05 mL of brain homogenate was used for the reaction mixture. In the homogenate, 0.05 M phosphate buffer, pH 7.0 (1.95 mL), and 0.019 M hydrogen peroxide (1.0 mL) were added for the final volume to make up the 3 mL. The absorbance of the reaction mixture was calculated at 240 nm after a 60-s interval for 2 min. The catalase activity was expressed in μmol/mg protein.
Estimation of plasma levels of phenobarbital
The levels of phenobarbital were estimated via the high-performance liquid chromatography analysis. Animals were sacrificed at two different time points, namely day 14 and day 28 after kindling, and blood was collected retroorbitally. The elution of phenobarbital was done from a reversed-phase column with an optimum temperature of 50°C. The mobile phase used consisted of 19/81 by the vol acetonitrile/phosphate buffer and the flow rate was set at 3.0 mL/min. The absorption of phenobarbital occurred at 195 nm and its quantity was estimated from the respective peak height. The sample injection volume was 10 μL. The time required for each analysis was about 14 min (Moriyama, 1999).
Histopathology of rat brain
The brain tissues were stored in 10% neutral formalin for the histopathological analysis. The brain tissues were processed by treating them with various concentrations of alcohol to remove the water from the tissues. The brain sections were then embedded in a molten wax and thin sections were cut (5 μm) and fixed on a glass slide. Then, it was stained with hematoxylin and eosin dye. Bright-field images were taken using an Olympus upright microscope.
The neuronal injury score was determined according to the following criteria: normal=no injury or rare isolated apoptotic neuron was given the score of 0; Rare neuronal injury less than 5 clusters=Score 1; The occasional neuronal injury (5-15 clusters)=Score 2; The frequent neuronal injury (>15 clusters)=Score 3; Diffuse neuronal injury=Score 4 (Wyler, 1992).
Statistical analysis
All data were expressed as Mean±SD. The quantitative parameters, such as seizure score and oxidative stress parameters were evaluated using a one-way analysis of variance (ANOVA) and the Bonferroni post hoc analysis. The unpaired student t-test was performed to compare the plasma concentration of different doses of PB. P<0.05 was considered statistically significant.
3. Results
Seizure scoring in PTZ kindled rats after the administration of different doses of phenobarbital for different periods
The rats in the saline as well as the CMC-treated groups demonstrated 0 seizure score throughout the 4-week kindling period. In the PTZ-treated group (30 mg/kg), the seizure score increased from 2.67±0.44 to 3.3±0.57 during the study period (Figure 2). Kindled rats treated with PB (20 mg/kg) showed no significant decline in the seizure score at the end of day 14 as well as day 28 of the study period. The phenobarbital 40 mg/kg (P<0.05) and 60 mg/kg (P<0.01) showed a significant reduction in the seizure score at the end of day 14 in the PTZ kindled animals. However, the chronic treatment of PB at the dose of 40 mg/kg and 60 mg/kg along with PTZ administration produced resistance as the seizure scores were found to be increased at the end of day 28 (P<0.05) (Figure 2).
Effects of phenobarbital treatment on reduced glutathione levels
The glutathione levels in rat brains were remarkably higher (P<0.05) in the PB 60 mg/kg treated group compared to the PTZ treated group and no significant effects were observed at PB dose 20 mg/kg and 40 mg/kg at the end of day 14. At the end of day 28 of PB treatment, no significant effects were observed in glutathione levels in all three treatment groups as compared to PTZ treated group (Figure 3).
Effects of phenobarbital treatment on superoxide dismutase levels
The level of SOD in rat brain was significantly reduced (P<0.05) in the PTZ treatment group as compared to vehicle, saline, and treated groups at the end of both 14 and 28 days of PB treatment. The decrease in the SOD levels was alleviated (P<0.05) with the treatment of PB (40 and 60 mg/kg) at the end of day 14. However, the chronic administrations of PB along with PTZ for 28 days in the kindled rats lead to a reduction in the levels of SOD (Figure 4).
Effect of phenobarbital treatment on catalase
The catalase levels in rat brains were significantly decreased (P<0.05) in PTZ treated group as compared to both the vehicle and saline-treated groups. The catalase levels were significantly increased (P<0.05) with PB treatment (40 and 60 mg/kg) dose on day 14. The treatment of PB (40 and 60 mg/kg) reduced the brain catalase levels as compared to the normal control group at the end of day 28 of PB treatment. However, this decrease in CAT level was not statistically significant (Figure 5). The brain catalase levels of phenobarbital 20 mg/kg group showed no significant effects when compared to PTZ control at day 28 of PB+PTZ treatment.
Effect on plasma levels of phenobarbital
The concentration of different doses of PB (20, 40, and 60 mg/kg) was measured in the rat’s blood at the end of day 14 day 28 of treatment with PB. The PB treatment at the dose of 20 mg/kg showed no significant increment in its plasma at the end of 28 days. However, a highly significant increase (P<0.01) in the PB levels was observed with the treatment of 40 mg/kg at the end of day 28 as compared to day 14 of PB treatment in PTZ kindled rats. The plasma levels of PB were significantly increased after 28 days of treatment at the dose of 60 mg/kg as compared to PTZ kindled rats. Moreover, the plasma concentration of PB was remarkably higher (P<0.05) in the 60 mg/kg PB treatment group in contrast to the 20 mg/kg PB treatment group at the end of day 28 (Table 1).
Neuronal injury score
The PTZ-treated group showed a significantly higher (P<0.001) neuronal injury score in comparison to the normal saline control group. The treatment of PB (40 and 60 mg/kg) significantly reduced (P<0.01) the neuronal injury score as compared to the PTZ-treated group at the end of day 14. However, the combination of PTZ and chronic administration of phenobarbital for 28 days increased the seizure severity and neuronal injury score leading to PB resistance. Thus, it was observed that the treatment of PB (40 and 60 mg/kg) reversed the decrease (P<0.05) in neuronal injury score at the end of day 28 when compared to day 14 of the PB+PTZ treatment period (Figure 6).
The PTZ-treated group showed a diffuse neuronal injury as compared to the normal saline group showing normal neurons with rare neuronal injury (Figure 7). The treatment of PB at 20 mg/kg dose showed frequent neuronal injury with (>15 clusters) at the end of 14 as well as 28 days of the study period. The occasional neuronal injury with (5-15 clusters) of neurons was observed with treatment of PB (40 and 60 mg/kg) at the end of day 14 of the study period. However, at the end of day 28, frequent neuronal injury with (>15 clusters) was observed with 40 and 60 mg/kg doses of PB treatment.
4. Discussion
This is the first study to elucidate and report the different parameters studied on PTZ-kindled Wistar rats at two different time points with three different doses of PB to select the dose at which maximum resistance occurs within a chronic period. The current research work reveals that phenobarbital treatment at 20 mg/kg dose showed no protective effect on seizure severity at any time point. The PB administration at both 40 mg/kg and 60 mg/kg doses showed an initial decrease in the seizure score and later developed resistance with an increase in seizure severity. This might be due to an increase in P-glycoprotein which is triggered by both seizure and the drug effect, thus leading to refractoriness (Jing et al., 2010). Moreover, drug tolerance is also one of the phenomena which leads to resistance. For example, phenobarbital undergoes pharmacokinetic tolerance which is attributed to its fast metabolism rate (Gay et al., 1983). Hence, in the current paper dose escalation (20 mg, 40 mg, 60 mg) method was used to combat tolerance at two (day 14 and day 28) time intervals. Increased dose helps in maintaining the same antiepileptic efficacy as observed earlier with low-dose treatment. The above-described tolerance is detected through plasma-level monitoring (Loscher & Schmidt, 2006). Following previous studies, a similar increase in the trend of PB levels in plasma has been observed at day 14 and day 28 in our research (Jing et al., 2010). However, one preclinical study on the spontaneous recurrent seizures model of drug-resistant epilepsy has also reported no differentiable comparison between plasma levels of PB responders and non-responders’ rodents (Brandt et al., 2004). PB epileptic brain is more prone to adverse effects because of the permanent alteration imprinted in the brain during epileptogenesis. There are studies reporting symptoms of sleepiness, dizziness, altered behavior, and depression as associated side effects among people with epilepsy taking PB at higher doses (Zhang et al., 2011). Therefore, to avoid the associated side effects 40 mg/kg was advised over 60 mg/kg, as the perfect dose of PB to induce resistance model which can easily be extrapolated in the clinical scenario.
The present pharmaco-resistant animal model has an advantage over conventional amygdala kindling and spontaneous recurrent seizures followed by PB and phenytoin treatment. It is less time-consuming and labor-intensive and could screen compounds over a short period. Moreover, no surgical expertise and electrode implants are required in the current animal model, which further decreases the chances of animal mortality. The major difference between our model and another promising pharmaco-resistance model is different in the study period. Our research model was a 56-day chronic study which is better suited to studying drug-resistant mechanisms. Additionally, chronic brain alteration can change the pharmacological response that cannot be visualized in acute models (Löscher, 2017).
Oxidative stress due to free radicals plays a pivotal role in the neurobiology of epilepsy. Recurrent seizures lead to the activation of the oxidative defense system and the production of free radicals (Shin et al., 2011). Further, the activation of free radicals leads to alteration in the levels of endogenous antioxidant enzymes, lipids, and expression of DNA (Ergul Erkec & Arihan, 2015). Numerous studies have depicted the involvement of certain endogenous (glutathione, catalase, superoxide dismutase) enzymes with neurodegenerative disorders (Cardenas-Rodriguez, 2013). Previous evidence supports the relationship between oxidative stress and mechanisms that lead to drug–resistance (Lorigados Pedre et al., 2018). The present study showed that the levels of antioxidant enzymes (glutathione, catalase, superoxide dismutase) in the PB 40 and 60 mg/kg treatment groups were decreased at the end of 28 days indicating the increase in oxidative stress, which depicts its positive correlation with drug-resistance epilepsy.
Previous reports have suggested that the majority of PB-resistant rats lead to major hippocampal damage (Bethmann, 2007). Based on preclinical findings it has been proposed that patients with uncontrollable seizures will have severe hippocampal damage as compared to patients with controlled epilepsy (Sloviter, 1994). Such clinical studies have found progressive damage to the hippocampus of patients with drug-resistant epilepsy (Kalviainen, 1998). Similar findings have been observed in our study where the combination of PTZ and chronic administration of the PB group has shown an increase in the neuronal injury score at the end of 28 days contributing to the process of epileptogenesis and the development of the model of drug resistance.
5. Conclusion
Drug resistance is the most challenging problem in the management of epilepsy. Therefore, it is necessary to understand the multifactorial mechanisms of epileptogenesis and develop animal models that target epileptogenesis for the better management of refractory epilepsy. Further, PTZ+PB (40 mg/kg) has shown the best results and it could be used to develop the refractory model of epilepsy and also to screen and study the various combinations of drugs that would help prevent and treat the resistance epilepsy.
Ethical Considerations
Compliance with ethical guidelines
All the experiments were performed according to the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), India. The study was approved by the Institutional Animal Ethics Committee and Institutional Biosafety Committee (No.: JSSCP/IAEC/OT/M.PHARM/PH.COLOGY/04/17-18).
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
Conceptualization: Bikash Medhi; Methodology: Nitika Garg, Amitava Chakrabarti, Bikash Medhi; Formal analysis and investigation: Surabhi Thapliyal, Nitika Garg; Writing- original draft preparation: Surabhi Thapliyal, Nitika Garg and Rupa Joshi; Review and editing: Nitika Garg, Rupa Joshi, Amitava Chakrabarti and Bikash Medhi; Supervision: Amitava Chakrabarti and Bikash Medhi.
Conflict of interest
The authors declared no conflict of interest.
Acknowledgments
The authors acknowledge the support from Experimental Pharmacology Laboratory (EPL) and PGIMER Chandīgarh, for providing all the experimental facilities to conduct the research.
References
Bethmann, K., Brandt, C., & Löscher, W. (2007). Resistance to phenobarbital extends to phenytoin in a rat model of temporal lobe epilepsy. Epilepsia, 48(4), 816-826. [DOI:10.1111/j.1528-1167.2007.00980.x] [PMID]
Brandt, C., Volk, H. A., & Löscher, W. (2004). Striking differences in individual anticonvulsant response to phenobarbital in rats with spontaneous seizures after status epilepticus. Epilepsia, 45(12), 1488–1497. [DOI:10.1111/j.0013-9580.2004.16904.x] [PMID]
Cardenas-Rodriguez, N., Huerta-Gertrudis, B., Rivera-Espinosa, L., Montesinos-Correa, H., Bandala, C., & Carmona-Aparicio, L., et al (2013). Role of Oxidative stress in refractory epilepsy: Evidence in patients and experimental models. International Journal of Molecular Sciences, 14(1), 1455–1476. [DOI:10.3390/ijms14011455] [PMID]
De Sarro, A., Naccari, F., & De Sarro, G. (1999). Enhanced susceptibility of pentylenetetrazole kindled mice to quinolone effects. International Journal of Antimicrobial Agents, 12(3), 239–244. [DOI:10.1016/S0924-8579(99)00067-9] [PMID]
Ellman, G. L. (1959). Tissue sulfhydryl groups. Archives of Biochemistry and Biophysics, 82(1), 70–77. [DOI:10.1016/0003-9861(59)90090-6] [PMID]
Ergul Erkec, O., & Arihan, O. (2015). Pentylenetetrazol kindling epilepsy model. Turkish Epilepsy Society, 21(1), 6-12 [DOI:10.5505/epilepsi.2015.08108]
Gay, M. H., Ryan, G. P., Boisse, N. R., & Guarino, J. J. (1983). Phenobarbital tolerance and physical dependence: Chronically equivalent dosing model. European Journal of Pharmacology, 95(1-2), 21–29. [DOI:10.1016/0014-2999(83)90263-7] [PMID]
Greenwald, R. A. (2017). Handbook methods for oxygen radical research. Boca Raton: CRC Press. [Link]
Ilhan, A., Iraz, M., Gurel, A., Armutcu, F., & Akyol, O. (2004). Caffeic acid phenethyl ester exerts a neuroprotective effect on CNS against pentylenetetrazol-induced seizures in mice. Neurochemical Research, 29(12), 2287–2292. [DOI:10.1007/s11064-004-7038-y] [PMID]
Jing, X., Liu, X., Wen, T., Xie, S., Yao, D., & Liu, X., et al (2010). Combined effects of epileptic seizure and phenobarbital induced overexpression of P-glycoprotein in brain of chemically kindled rats. British Journal of Pharmacology, 159(7), 1511–1522. [DOI:10.1111/j.1476-5381.2009.00634.x] [PMID]
Kälviäinen, R., Salmenperä, T., Partanen, K., Vainio, P., Riekkinen, P., & Pitkänen, A. (1998). Recurrent seizures may cause hippocambal damage in temporal lobe epilepsy. Neurology, 50(5), 1377–1382. [DOI:10.1212/WNL.50.5.1377] [PMID]
Kono, Y. (1978). Generation of superoxide radical during autoxidation of hydroxylamine and an assay for superoxide dismutase. Archives of Biochemistry and Biophysics, 186(1), 189–195. [DOI:10.1016/0003-9861(78)90479-4] [PMID]
Kumar, B., Medhi, B., Modi, M., Saikia, B., Attri, S. V., & Patial, A. (2018). A mechanistic approach to explore the neuroprotective potential of zonisamide in seizures. Inflammopharmacology, 26(4), 1125–1131. [DOI:10.1007/s10787-018-0478-9] [PMID]
Kwan, P., Arzimanoglou, A., Berg, A. T., Brodie, M. J., Allen Hauser, W., & Mathern, G., et al. (2010). Definition of drug resistant epilepsy: Consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia, 51(6), 1069–1077. [DOI:10.1111/j.1528-1167.2009.02397.x] [PMID]
Lalitha, S., Minz, R. W., & Medhi, B. (2018). Understanding the controversial drug targets in epilepsy and pharmacoresistant epilepsy. Reviews in The Neurosciences, 29(3), 333–345. [DOI:10.1515/revneuro-2017-0043] [PMID]
Lorigados Pedre, L., Gallardo, J. M., Morales Chacón, L. M., Vega García, A., Flores-Mendoza, M., & Neri-Gómez, T., et al. (2018). Oxidative stress in patients with drug resistant partial complex seizure. Behavioral Sciences (Basel, Switzerland), 8(6), 59. [PMID]
Löscher, W. (2017). Animal models of drug-refractory epilepsy. In: A. Pitkänen, P. S. Buckmaster, & S. L. Moshé (Es.), Models of seizures and epilepsy (pp. 43-60). Massachusetts: Academic Press [DOI:10.1016/B978-0-12-804066-9.00051-1]
Löscher, W. (2007). The pharmacokinetics of antiepileptic drugs in rats: Consequences for maintaining effective drug levels during prolonged drug administration in rat models of epilepsy. Epilepsia, 48(7), 1245–1258. [DOI:10.1111/j.1528-1167.2007.01093.x] [PMID]
Löscher, W., & Schmidt, D. (2006). Experimental and clinical evidence for loss of effect (tolerance) during prolonged treatment with antiepileptic drugs. Epilepsia, 47(8), 1253-1284. [DOI:10.1111/j.1528-1167.2006.00607.x] [PMID]
Löscher, W. (1997). Animal models of intractable epilepsy. Progress in Neurobiology, 53(2), 239–258. [DOI:10.1016/S0301-0082(97)00035-X] [PMID]
Löscher, W., & Hönack, D. (1989). Comparison of the anticonvulsant efficacy of primidone and phenobarbital during chronic treatment of amygdala-kindled rats. European Journal of Pharmacology, 162(2), 309–322. [DOI:10.1016/0014-2999(89)90294-X] [PMID]
McNamara, J. O., Byrne, M. C., Dasheiff, R. M., & Fitz, J. G. (1980). The kindling model of epilepsy: A review. Progress in Neurobiology, 15(2), 139–159. [DOI:10.1016/0301-0082(80)90006-4] [PMID]
Moriyama, M., Yamashita, S., Domoto, H., Furuno, K., Araki, H., & Gomita, Y. (1999). Determination of plasma phenobarbital concentration by high-performance liquid chromatography in rat offspring. Journal of Chromatography. B, Biomedical Sciences and Applications, 723(1-2), 301–305. [DOI:10.1016/S0378-4347(98)00534-9] [PMID]
Pavlova, T., Stepanichev, M., & Gulyaeva, N. (2006). Pentylenetetrazole kindling induces neuronal cyclin B1 expression in rat hippocampus. Neuroscience Letters, 392(1-2), 154–158.[DOI:10.1016/j.neulet.2005.09.021] [PMID]
Saha, L., & Chakrabarti, A. (2014). Understanding the anti-kindling role and its mechanism of Resveratrol in Pentylenetetrazole induced-kindling in a rat model. Pharmacology, Biochemistry, and Behavior, 120, 57–64. [DOI:10.1016/j.pbb.2014.01.010] [PMID]
Sato, M., Racine, R. J., & McIntyre, D. C. (1990). Kindling: Basic mechanisms and clinical validity. Electroencephalography and Clinical Neurophysiology, 76(5), 459-472. [DOI:10.1016/0013-4694(90)90099-6]
Shin, E. J., Jeong, J. H., Chung, Y. H., Kim, W. K., Ko, K. H., & Bach, J. H., et al. (2011). Role of oxidative stress in epileptic seizures. Neurochemistry International, 59(2), 122–137. [DOI:10.1016/j.neuint.2011.03.025] [PMID]
Singh, E., Pillai, K. K., & Mehndiratta, M. (2014). Characterization of a lamotrigine-resistant kindled model of epilepsy in mice: Evaluation of drug resistance mechanisms. Basic & Clinical Pharmacology & Toxicology, 115(5), 373–378. [DOI:10.1111/bcpt.12238] [PMID]
Singh, T., Mishra, A., & Goel, R. K. (2021). PTZ kindling model for epileptogenesis, refractory epilepsy, and associated comorbidities: Relevance and reliability. Metabolic Brain Disease, 36(7), 1573-1590. [DOI:10.1007/s11011-021-00823-3] [PMID]
Singh, T., Kaur, T., & Goel, R. K. (2017). Ferulic acid supplementation for management of depression in epilepsy. Neurochemical Research, 42(10), 2940–2948. [DOI:10.1007/s11064-017-2325-6] [PMID]
Singh, T., Kaur, T., & Goel, R. K. (2017). Adjuvant quercetin therapy for combined treatment of epilepsy and comorbid depression. Neurochemistry International, 104, 27–33. [DOI:10.1016/j.neuint.2016.12.023] [PMID]
Singh, T., & Goel, R. K. (2017). Adjuvant neuronal nitric oxide synthase inhibition for combined treatment of epilepsy and comorbid depression. Pharmacological Reports: PR, 69(1), 143–149. [DOI:10.1016/j.pharep.2016.10.001] [PMID]
Singh, T., & Goel, R. K. (2016). Adjuvant indoleamine 2,3-dioxygenase enzyme inhibition for comprehensive management of epilepsy and comorbid depression. European Journal of Pharmacology, 784, 111–120. [DOI:10.1016/j.ejphar.2016.05.019] [PMID]
Sloviter R. S. (1994). The functional organization of the hippocampal dentate gyrus and its relevance to the pathogenesis of temporal lobe epilepsy. Annals of Neurology, 35(6), 640–654.[DOI:10.1002/ana.410350604] [PMID]
Wyler, A. R., Curtis Dohan, F., Schweitzer, J. B., & Berry, A. D. (1992). A grading system for mesial temporal pathology (hippocampal sclerosis) from anterior temporal lobectomy. Journal of Epilepsy, 5(4), 220-225. [DOI:10.1016/S0896-6974(05)80120-3]
Zhang, L. L., Zeng, L. N., & Li, Y. P. (2011). Side effects of phenobarbital in epilepsy: A systematic review. Epileptic Disorders: International Epilepsy Journal with Videotape, 13(4), 349–365. [DOI:10.1684/epd.2011.0444] [PMID]
Type of Study: Original |
Subject:
Behavioral Neuroscience
Received: 2021/12/20 | Accepted: 2022/03/9 | Published: 2023/09/1
Received: 2021/12/20 | Accepted: 2022/03/9 | Published: 2023/09/1
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
