

Volume 11, Issue 6 (November & December 2020)
BCN 2020, 11(6): 781-794 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Eslami Gharaati M, Nahavandi A, Baluchnejad Mojarad T, Roghani M. Diabetic Encephalopathy Affecting Mitochondria and Axonal Transport Proteins. BCN 2020; 11 (6) :781-794
URL: http://bcn.iums.ac.ir/article-1-1396-en.html
URL: http://bcn.iums.ac.ir/article-1-1396-en.html



1- Department of Physiology, School of Medicine, Iran University of Medical Science, Tehran, Iran.
2- Departmentof Physiology, Neurophysiology Research Center, Shahed University, Tehran, Iran.
2- Departmentof Physiology, Neurophysiology Research Center, Shahed University, Tehran, Iran.
Keywords: Diabetes mellitus type 1, Mitochondrial encephalopathy, Axonal transport, Mitochondria, KIF5b protein, Dynein
Full-Text [PDF 917 kb]
| Abstract (HTML)
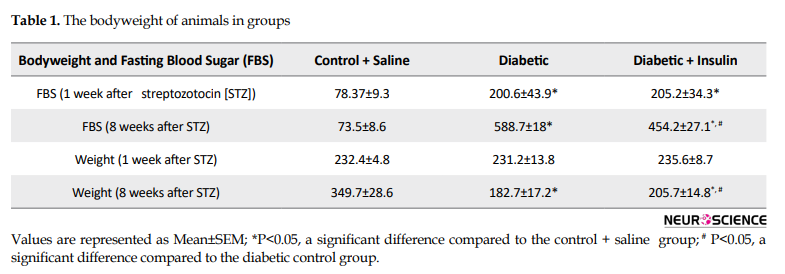
Significant impairment in weight gain occurred in the diabetic rats comparing to the control rats in the eighth week after the induction of diabetes (F2,21=151.06, P=0.0001). Also in the diabetic group, FBS significantly increased compared to the control group (F2,21=305.57, P=0.0001). Insulin injection for 8 weeks improved weight gain in diabetic rats. Animals’ weight in the diabetic + insulin was significantly higher than that in the diabetic rats (P<0.05). FBS was significantly lower than that in the diabetic rats (P<0.05) (Table 1).
3.2. Behavioral tests
3.2.1. Elevated Plus Maze (EPM)
The number of entries to open arm and time spent in open arms were considered as anxiety indices and presented as a percentage. Time spent in open arms (%) was significantly decreased (F2,18=4.88, P=0.02) (Figure 1).
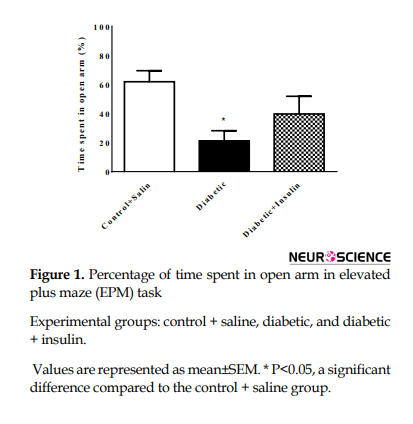
This index decreased in diabetic rat compared to that in the control group (control + saline = 61.88±20.14, diabetic=21.18±18.49, P<0.05). Whereas the number of open arm entrance (%) was not changed in groups (F2,18=2.26, P=0.1324) (Figure 2).
.PNG)
3.2.2. Y-maze
Y-maze test was performed 48 hours after EPM. The percentage of alternation behavior 8 weeks after the induction of diabetes was significantly changed (F2,18=3.60, P=0.048) (Figure 3).
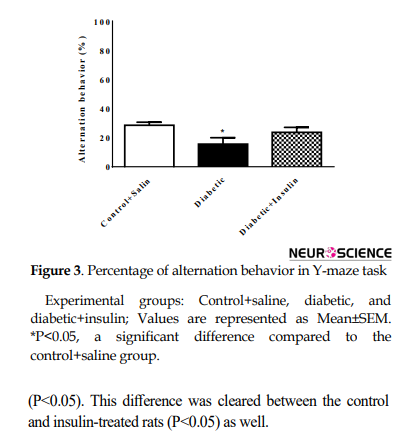
This index decreased in diabetic rats compared to the control group (control + saline =28.57±5.53, diabetic=15.57±11.77, P<0.05).
3.2.3. Passive Avoidance Learning (PAL)
This test was performed 48 h after the Y-maze task. The initial latency was recorded when the rats entered the dark compartment. In all three groups, there was no significant change in IL (F2,18=0.686, P=0.515) (Figure 4).
.PNG)
The step-through latency (maximum 480 s) was measured and recorded as the index for passive avoidance learning. STL index was significantly changed (F2,18=7.31, P=0.0047) (Figure 5).
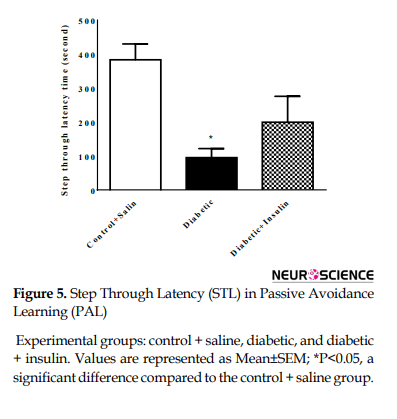
STL in the diabetic group decreased compared to that in the control group (control + saline =384±123.94, diabetic=95.28±70.77, P<0.05).
3.2.4. Gene expression of KIF5b and Dynein by real-time PCR
Real-time PCR was carried out to reveal KIF5b and dynein gene expression (Figure 6).
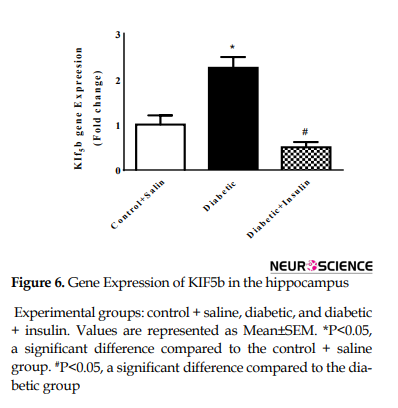
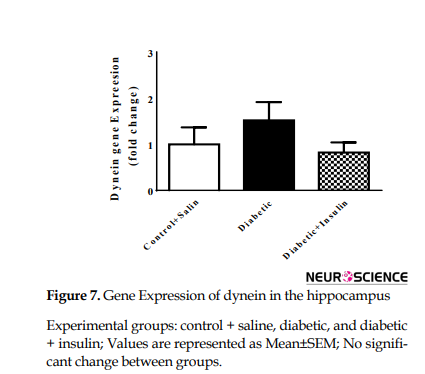
Gene expression of KIF5b dramatically changed after 8 weeks of diabetes induction (F2,9=20.41, P=0.0005). Actually, KIF5b mRNA levels were significantly increased compared to the control group (control + saline = 1±0.4, diabetic = 2.25±0.47, P<0.05). Insulin injection for 8 weeks significantly decreased expression of KIF5b compared with diabetic rats (diabetic = 2.25±0.4796, diabetic + insulin=0.5±0.23, P<0.05) (Figure 6). No significant changes were observed in dynein mRNA levels in different groups (F2,9=1.86, P =0.2096) (Figure 7).
3.2.5. Mitochondrial membrane potential by rhodamine 123 probe
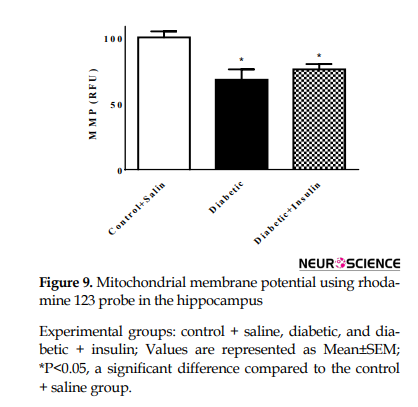
The Mitochondrial Membrane Potential (MMP) of cells was assessed by monitoring the uptake of cationic dye rhodamine 123. The results of MMP showed a significant change in different groups (F2,18= 8.557, P=0.0024) (Figure 8).
In the diabetic group, MMP decreased 8 weeks after induction of diabetes compared to that in the control group (control + saline=100.1±12.14, diabetic = 68.05±20.64, P<0.05) and there was a significant difference between the control group and insulin injected groups (control + saline=100.1±12.14, diabetic + insulin=75.88±10.58, P<0.05).
3.2.6. Biochemical parameters
Serum level of insulin and the percentage of glycosylated hemoglobin (HgbA1c %) were measured by using ELISA kits. Mean±SEM of HgbA1c (%) and serum insulin are given in Table 2 Results of HgbA1c percentage showed a significant difference between the groups (F2,18=95.99, P=0.0001).
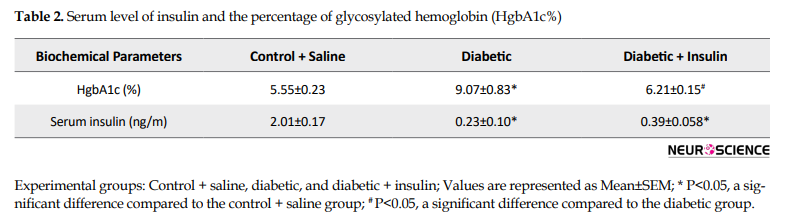
Diabetic animals revealed higher glycosylated hemoglobin compared to age-matched controls (P<0.05). A significant difference was observed between the diabetic and insulin-treated group (P<0.05).
The serum level of insulin was significantly different between the groups (F2,12= 316.3, P=0.0001). Insulin levels in diabetic rats were significantly lower than control rats (P<0.05). This difference was cleared between the control and insulin-treated rats (P<0.05) as well.
4. Discussion
Diabetic encephalopathy is referred to as any cognitive dysfunction following diabetes mellitus. This is why this condition is called type 3 DM (T3DM) or Alzheimer disease (Steen et al., 2005; Rivera et al., 2005; de la Monte, & Wands, 2008; Leszek et al., 2017). On the other hand, Mitochondrial Diabetes (MD) appears after the activation of mutant mitochondrial DNA, which is age-dependent. MD is the combination of diabetes and cognitive deficit (Maassen et al., 2004; Maassen, Janssen, & Hart, 2005). In this research, we showed the correlation between diabetic encephalopathy, dysfunctional mitochondria, and change in the expression of axonal transport protein kinesin (KIF5b).
We showed short-term memory impairment and retrieval deficit in combination with anxiety-like behavior following T3DM. To measure anxiety-like behavior in rodents, the elevated plus maze is an accepted test (Pellow, Chopin, File, & Briley, 1985). Our findings on EPM showed a decrease in time spent in open arm, in the diabetic group, compared with the control and or insulin-injected groups. In 2007, Miyata showed an increase of anxiety-like behavior following STZ-induced T1DM in rodents (Miyata et al., 2007).
Another study has revealed that N-acetylcysteine (NAC) is protective against anxiety-like behavior in the EPM test of diabetic rats (Kamboj, Chopra, & Sandhir, 2008). Behavioral changes due to diabetic encephalopathy are not only dependent on the insulin metabolism cascade impairment, but also because of decreased serotonin levels in the central nervous system (Zalsman et al., 2006, Pittenger & Duman, 2008). On the other hand, hyperglycemia induces neurodegeneration and pathologic behavior and cognition. In the diabetic + insulin group, insulin has possibly prevented behavioral deficits. Our findings in the passive avoidance learning task showed different degrees of learning and memory impairment in type 3 diabetes. Actually, in the shuttle box, the significant decrease in Step-Through Latency (STL) in T3DM is referred to impairment of memory retention and consolidation. Scientists have reported the role of insulin on the improvement of memory in DM, which is in agreement with our results (diabetic + insulin group showing an increase in STL by insulin administration). Following prolonged DM, the impaired hippocampal plasticity may damage learning and memory processing (Flood, Mooradian, & Morley, 1990; Stewart & Liolitsa, 1999; Stranahan et al., 2008). Scientists have found that anxiety level and cognitive dysfunction is directly correlated to diabetes duration in young rats (Rajashree, Kholkute, & Goudar, 2011). Memory and learning deficit and even behavioral changes are among obvious manifestations of diabetic encephalopathy, as insulin and c-peptide are key factors for synaptic plasticity and cognition (Sima et al., 2009; McNay & Recknagel, 2011). Scientists have revealed the role of ICV streptozotocin in the induction of cognitive dysfunction, which has been reversed after insulin infusion (Guo et al., 2017). Physiologic mitochondrial function is dependent on its intact transport process (Chada & Hollenbeck, 2004; Russo et al., 2009). Low insulin and c-peptide levels may disrupt normal cerebral metabolism and function sequentially (de la Monte & Wands, 2008; Sima et al., 2009).
Although our finding was in favor of the late course of T1DM or T3DM, the hypoglycemic course is related to cerebral malfunction is not clear. However, the chronicity of T1DM is correlated with encephalopathy (Garg, Bonanome, Grundy, Zhang, & Unger, 1988; Musen et al., 2006).
Natural flavonoids have been effective in the reversal of anxiety-like behavior detected in the EPM test in diabetic mice (Damian et al., 2014; Tang et al., 2015). Insulin plays a critical role in hippocampal memory processing and hyperglycemia is responsible for the cognitive deficit and neuronal damage following DM (Abbatecola et al., 2006; Strachan et al., 2011).
In rhodamine monitoring of hippocampal mitochondrial membrane potential, we found a decreased level of mitochondrial staining. Mitochondrial function can be monitored by MMP (Figure 9).
The normal detectable electrochemical gradient of mitochondrial membrane refers to ATP production (Hafner, Brown, & Brand, 1990; Fontaine, Devin, Rigoulet, & Leverve, 1997). Rhodamine 123 transmembrane distribution reveals membrane potential strength (Emaus, Grunwald, & Lemasters, 1986; Huang, Camara, Stowe, Qi, & Beard, 2007). Following any mitochondrial and cellular damage, there will be abnormal or undetectable MMP and ATP production and neuronal apoptosis (Letai 2006; de la Monte, 2012). Even low MMP represents apoptosis (Ding, Han, Zhu, Chen, & D’Ambrosio, 2005). Electrophoretic mobility of mitochondria is directly related to its permeability and staining specifies (Emauset al., 1986; Bunting, 1992). In 2017, scientists have shown that normal mitochondrial metabolism potentially prevents from decreased age-dependent neurogenesis in the hippocampus (Beckervordersandforth et al., 2017). Besides mitochondrial count and morphology, mitochondrial metabolism also depends on the dynamic movement and distribution of these organelles, which is essential to maintain normal ATP synthesis (Lu et al., 2004).
Our study showed a higher expression of KIF5b, a member of the kinesin superfamily. KIF5b has a key role in both mitochondrial translocation and distribution. KIF5b depletion will stop normal mitochondrial transport. In 2011 and for the first time, scientists showed the vital role of kinesin protein in mitochondrial transport of hippocampal neurons (Brickley & Anne Stephenson, 2011).
In 2013, scientists demonstrsted increased KIF5b gene expression in cultured hippocampal neurons of diabetic rats with no alterations in dynein gene expression (Baptista, Pinto, Elvas, Almeida, & Ambrósio, 2013). Dynein is not only responsible for retrograde translocation of mitochondria, but responsible for the movement of other motor adaptors (King & Schroer, 2000, Pilling, Horiuchi, Lively, & Saxton, 2006). Although some kinesins are also involved in retrograde transport (Hirokawa, Noda, Tanaka, & Niwa, 2009).
In this study, we found that diabetes alters the KIF5b motor protein by increasing KIF5b gene expression levels in the hippocampus 8 weeks after the induction of diabetes and the anterograde transport of mitochondria may be impaired in the hippocampus. As a consequence, we suggested that a decrease in the number of synaptic vesicles and density may ultimately account for changes in synaptic transmission and mitochondrial transport in the hippocampus. In 2014, the effect of T1DM (duration of 2 to 8 weeks) on rat retinal distribution of dynein, KIF5b, and KIF1a in retinal tissue was confirmed (Baptista et al., 2014), which was considered as the possible factor for diabetic retinal neurodegeneration.
In this study, we found no change in dynein heavy chain expression. Although to our knowledge there is still no report about encephalopathy and dynein gene expression, it may be a more chronic type of T3DM model (longer than 8 weeks) that could reveal different findings. While IHC, as a qualitative method and western blotting as a quantitative one, shows protein end product and complete study, when mRNA production is impaired no protein will be found.
In conclusion, this study revealed the correlation between diabetic encephalopathy, mitochondrial dysfunction, and kinesin gene overexpression. We suggest that abnormal translocation of mitochondria and its malfunction induced by kinesin gene overexpression may result in diabetic encephalopathy.
Ethical Considerations
Compliance with ethical guidelines
All ethical principles are considered in this article in accordance with Physiology Research Center and Cellular and Molecular Research Center of Iran University of Medical Sciences.
Funding
This research did not receive any specific grant or budget from funding agencies in the public, commercial, or not-for-profit sectors.
Authors' contributions
All authors equally contributed to preparing this article.
Conflict of interest
The authors declared no conflict of interest to the publication of this article.
Acknowledgments
We would like to thank our colleagues from the Physiology Research Center and Cellular and Molecular Research Center of Iran University of Medical Sciences.
References
Abbatecola, A. M., Rizzo, M. R., Barbieri, M., Grella, R., Arciello, A., & Laieta, M. T., et al. (2006). Postprandial plasma glucose excursions and cognitive functioning in aged type 2 diabetics. Neurology, 67(2), 235-40. [DOI:10.1212/01.wnl.0000224760.22802.e8] [PMID]
Abebe, W., Harris, K. H., & MacLeod, K. M. (1990). Enhanced contractile responses of arteries from diabetic rats to alpha 1-adrenoceptor stimulation in the absence and presence of extracellular calcium. Journal of Cardiovascular Pharmacology, 16(2), 239-48. [DOI:10.1097/00005344-199008000-00010] [PMID]
Arfa-Fatollahkhani, P., Nahavandi, A., Abtahi, H., Anjidani, S., Borhani, S., & Jameie, S. B., et al. (2017). The effect of luteinizing hormone reducing agent on anxiety and novel object recognition memory in gonadectomized rats. Basic and Clinical Neuroscience, 8(2), 113-9. [DOI:10.18869/nirp.bcn.8.2.113] [PMID] [PMCID]
Baloh, R. H., Schmidt, R. E., Pestronk, A., & Milbrandt, J. (2007). Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. The Journal of Neuroscience, 27(2), 422-30. [DOI:10.1523/JNEUROSCI.4798-06.2007] [PMID] [PMCID]
Baptista, F. I., Pinto, M. J., Elvas, F., Almeida R. D., & Ambrósio, A. F. (2013). Diabetes alters KIF1A and KIF5B motor proteins in the hippocampus. PLoS One, 8(6), e65515. [DOI:10.1371/journal.pone.0065515] [PMID] [PMCID]
Baptista, F. I., Pinto, M. J., Elvas, F., Martins, T., Almeida, R. D., & Ambrosio, A. F. (2014). Diabetes induces changes in KIF1A, KIF5B and dynein distribution in the rat retina: Implications for axonal transport. Experimental Eye Research, 127, 91-103. [DOI:10.1016/j.exer.2014.07.011] [PMID]
Barel, O., Malicdan, M. C. V., Ben-Zeev, B., Kandel, J., Pri-Chen, H., & Stephen, J., et al. (2017). Deleterious variants in TRAK1 disrupt mitochondrial movement and cause fatal encephalopathy. Brain, 140(3), 568-81. [DOI:10.1093/brain/awx002] [PMID] [PMCID]
Beckervordersandforth, R., Ebert, B., Schaffner, I., Moss, J., Fiebig, C., & Shin, J., et al. (2017). Role of mitochondrial metabolism in the control of early lineage progression and aging phenotypes in adult hippocampal neurogenesis. Neuron, 93(6), 1518. [DOI:10.1016/j.neuron.2017.03.008] [PMID] [PMCID]
Beckman, K. B., & Ames, B. N. (1998). The free radical theory of aging matures. Physiological Reviews, 78(2), 547-81. [DOI:10.1152/physrev.1998.78.2.547] [PMID]
Belkacemi, A., & Ramassamy, C. (2012). Time sequence of oxidative stress in the brain from transgenic mouse models of Alzheimer’s disease related to the amyloid-beta cascade. Free Radical Biology and Medicine, 52(3), 593-600. [DOI:10.1016/j.freeradbiomed.2011.11.020] [PMID]
Bessman, S. P., & Bessman, A. N. (1955). The cerebral and peripheral uptake of ammonia in liver disease with an hypothesis for the mechanism of hepatic coma. The Journal of Clinical Investigation, 34(4), 622-8. [DOI:10.1172/JCI103111] [PMID] [PMCID]
Brands, M. W., Bell, T. D., & Gibson, B. (2004). Nitric oxide may prevent hypertension early in diabetes by counteracting renal actions of superoxide. Hypertension, 43(1), 57-63. [DOI:10.1161/01.HYP.0000104524.25807.EE] [PMID]
Brickley, K., Anne Stephenson, F. (2011). Trafficking kinesin protein (TRAK)-mediated transport of mitochondria in axons of hippocampal neuron. The Journal of Biological Chemistry, 286(20), 18079-92. [DOI:10.1074/jbc.M111.236018] [PMID] [PMCID]
Bunting, J. R. (1992). Influx and efflux kinetics of cationic dye binding to respiring mitochondria. Biophysical Chemistry, 42(2), 163-75. [DOI:10.1016/0301-4622(92)85006-P]
Cai, D., & Liu, T. (2012). Inflammatory cause of metabolic syndrome via brain stress and NF-kappaB. Aging, 4(2), 98-115. [DOI:10.18632/aging.100431] [PMID] [PMCID]
Chada, S. R., & Hollenbeck, P. J. (2004). Nerve growth factor signaling regulates motility and docking of axonal mitochondria. Current Biology, 14(14), 1272-6. [DOI:10.1016/j.cub.2004.07.027] [PMID]
Connor, B., Young, D., Yan, Q., Faull, R. L. M., Synek, B., & Dragunow, M. (1997). Brain-derived neurotrophic factor is reduced in Alzheimer’s disease. Molecular Brain Research, 49(1-2), 71-81. [DOI:10.1016/S0169-328X(97)00125-3]
Damian, J. P., Acosta, V., Da Cuña, M., Ramirez, I., Oddone, N., & Zambrana, A., et al. (2014). Effect of resveratrol on behavioral performance of streptozotocin-induced diabetic mice in anxiety tests. Experimental Animals, 63(3), 277-87. [DOI:10.1538/expanim.63.277] [PMID] [PMCID]
de la Monte, S. M. (2012). Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Current Alzheimer Research, 9(1), 35-66. [DOI:10.2174/156720512799015037] [PMID] [PMCID]
de la Monte, S. M., & Wands, J. R. (2008). Alzheimer’s disease is type 3 diabetes-evidence reviewed. Journal of Diabetes Science and Technology, 2(6), 1101-13. [DOI:10.1177/193229680800200619] [PMID] [PMCID]
Ding, H., Han, C., Zhu, J., Chen, C. S., & D’Ambrosio, S. M. (2005). Celecoxib derivatives induce apoptosis via the disruption of mitochondrial membrane potential and activation of caspase 9. International Journal of Cancer, 113(5), 803-10. [DOI:10.1002/ijc.20639] [PMID]
Ding, J., Yu, H. L., Ma, W. W., Xi, Y. D., Zhao, X., & Yuan, L. H., et al. (2013). Soy isoflavone attenuates brain mitochondrial oxidative stress induced by beta-amyloid peptides 1-42 injection in lateral cerebral ventricle. Journal of Neuroscience Research, 91(4), 562-7. [DOI:10.1002/jnr.23163] [PMID]
Emaus, R. K., Grunwald, R., & Lemasters, J. J. (1986). Rhodamine 123 as a probe of transmembrane potential in isolated rat-liver mitochondria: Spectral and metabolic properties. Biochimica et Biophysica Acta (BBA) - Bioenergetics, 850(3), 436-48. [DOI:10.1016/0005-2728(86)90112-X]
Finsterer, J. (2006). Central nervous system manifestations of mitochondrial disorders. Acta neurologica Scandinavica, 114(4), 217-38. [DOI:10.1111/j.1600-0404.2006.00671.x] [PMID]
Flood, J. F., Mooradian, A. D., & Morley, J. E. (1990). Characteristics of learning and memory in streptozocin-induced diabetic mice. Diabetes, 39(11), 1391-8. [DOI:10.2337/diab.39.11.1391] [PMID]
Fontaine, E. M., Devin, A., Rigoulet, M., & Leverve, X. M. (1997). The yield of oxidative phosphorylation is controlled both by force and flux. Biochemical and Biophysical Research Communications, 232(2), 532-5. [DOI:10.1006/bbrc.1997.6317] [PMID]
Garg, A., Bonanome, A., Grundy, S. M., Zhang, Z. J., & Unger, R. H. (1988). Comparison of a high-carbohydrate diet with a high-monounsaturated-fat diet in patients with non-insulin-dependent diabetes mellitus. The New England Journal of Medicine, 319(13), 829-34. [DOI:10.1056/NEJM198809293191304] [PMID]
Guo, Z., Chen, Y., Mao, Y. F., Zheng, T., Jiang, Y., & Yan, Y., et al. (2017). Long-term treatment with intranasal insulin ameliorates cognitive impairment, tau hyperphosphorylation, and microglial activation in a streptozotocin-induced Alzheimer’s rat model. Scientific Reports, 7, 45971. [DOI:10.1038/srep45971] [PMID] [PMCID]
Haas, R. H., Parikh, S., Falk, M. J., Saneto, R. P., Wolf, N. I., & Darin N., et al. (2007). Mitochondrial disease: A practical approach for primary care physicians. Pediatrics, 120(6), 1326-33. [DOI:10.1542/peds.2007-0391] [PMID]
Hafner, R. P., Brown, G. C., & Brand, M. D. (1990). Analysis of the control of respiration rate, phosphorylation rate, proton leak rate and protonmotive force in isolated mitochondria using the ‘top-down’ approach of metabolic control theory. European Journal of Biochemistry, 188(2), 313-9. [DOI:10.1111/j.1432-1033.1990.tb15405.x] [PMID]
Hindfelt, B., Plum, F., & Duffy, T. E. (1977). Effect of acute ammonia intoxication on cerebral metabolism in rats with portacaval shunts. The Journal of Clinical Investigation, 59(3), 386-96. [DOI:10.1172/JCI108651] [PMID] [PMCID]
Hirokawa, N., Noda, Y., Tanaka, Y., & Niwa, Sh. (2009). Kinesin superfamily motor proteins and intracellular transport. Nature Reviews Molecular Cell Biology Volume, 10(10), 682-96. [DOI:10.1038/nrm2774] [PMID]
Hollenbeck, P. J., & Saxton, W. M. (2005). The axonal transport of mitochondria. Journal of Cell Science, 118(Pt 23), 5411-9. [DOI:10.1242/jcs.02745] [PMID] [PMCID]
Hritcu, L., Cioanca, O., & Hancianu, M. (2012). Effects of lavender oil inhalation on improving scopolamine-induced spatial memory impairment in laboratory rats. Phytomedicine, 19(6), 529-34. [DOI:10.1016/j.phymed.2012.02.002] [PMID]
Huang, M., Camara, A. K., Stowe, D. F., Qi, F., & Beard, D. A. (2007). Mitochondrial inner membrane electrophysiology assessed by rhodamine-123 transport and fluorescence. Annals of Biomedical Engineering, 35(7), 1276-85. [DOI:10.1007/s10439-007-9265-2] [PMID] [PMCID]
Kamboj, S. S., Chopra, K., & Sandhir, R. (2008). Neuroprotective effect of N-acetylcysteine in the development of diabetic encephalopathy in streptozotocin-induced diabetes. Metabolic Brain Disease, 23(4), 427. [DOI:10.1007/s11011-008-9104-7] [PMID]
King, S. J., & Schroer, T. A. (2000). Dynactin increases the processivity of the cytoplasmic dynein motor. Nature Cell Biology, 2(1), 20-4. [DOI:10.1038/71338] [PMID]
Leszek, J., Trypka, E., Tarasov, V. V., Ashraf, G. M., & Aliev, G. (2017). Type 3 diabetes mellitus: A novel implication of Alzheimers disease. Current Topics in Medicinal Chemistry, 17(12), 1331-5. [DOI:10.2174/1568026617666170103163403] [PMID]
Letai, A. (2006). Growth factor withdrawal and apoptosis: The middle game. Molecular Cell, 21(6), 728-30. [DOI:10.1016/j.molcel.2006.03.005] [PMID]
Lu, T., Pan, Y., Kao, S. Y., Li, C., Kohane, I., & Chan, J., et al. (2004). Gene regulation and DNA damage in the ageing human brain. Nature, 429(6994), 883-91. [DOI:10.1038/nature02661] [PMID]
Lulat, S. I., Yadav, Y. C., Balaraman, R., & Maheshwari, R. (2016). Antiurolithiatic effect of lithocare against ethylene glycol-induced urolithiasis in Wistar rats. Indian Journal of Pharmacology, 48(1), 78-82. [DOI:10.4103/0253-7613.174564] [PMID] [PMCID]
Lupien, S. B., Bluhm, E. J., & Ishii, D. N. (2003). Systemic insulin-like growth factor-I administration prevents cognitive impairment in diabetic rats, and brain IGF regulates learning/memory in normal adult rats. Journal of Neuroscience Research, 74(4), 512-23. [DOI:10.1002/jnr.10791] [PMID]
Maassen, J. A., ‘t Hart, L. M., van Essen, E., Heine, R. J., Nijpels, G., & Jahangir Tafrechi, R. S., et al. (2004). Mitochondrial diabetes: Molecular mechanisms and clinical presentation. Diabetes, 53(Suppl 1), S103-9. [DOI:10.2337/diabetes.53.2007.S103] [PMID]
Maassen, J. A., Janssen, G. M. C., & ‘t Hart, L. M. (2005). Molecular mechanisms of mitochondrial diabetes (MIDD). Annals of Medicine, 37(3), 213-21. [DOI:10.1080/07853890510007188] [PMID]
McNay, E. C., & Recknagel, A. K. (2011). Brain insulin signaling: A key component of cognitive processes and a potential basis for cognitive impairment in type 2 diabetes. Neurobiology of Learning and Memory, 96(3), 432-42. [DOI:10.1016/j.nlm.2011.08.005] [PMID] [PMCID]
Mingatto, F. E., Rodrigues, T., Pigoso, A. A., Uyemura, S. A., Curti, C., & Santos, A. C. (2002). The critical role of mitochondrial energetic impairment in the toxicity of nimesulide to hepatocytes. Journal of Pharmacology and Experimental Therapeutics, 303(2), 601-7. [DOI:10.1124/jpet.102.038620] [PMID]
Miyata, Sh., Yamada, N., Hirano, S., Tanaka, S. I., & Kamei, J. (2007). Diabetes attenuates psychological stress-elicited 5-HT secretion in the prefrontal cortex but not in the amygdala of mice. Brain Research, 1147, 233-9. [DOI:10.1016/j.brainres.2007.02.001] [PMID]
Mousavi, F., Eidi, A., Khalili, M., & Roghani, M. (2018). Metformin ameliorates learning and memory deficits in streptozotocin-induced diabetic rats. Journal of Basic and Clinical Pathophysiology, 6(1), 17-22. [DOI:10.22070/JBCP.2017.2782.1085]
Musen, G., Lyoo, I. K., Sparks, C. R., Weinger, K., Hwang, J., & Ryan, C. M., et al. (2006). Effects of type 1 diabetes on gray matter density as measured by voxel-based morphometry. Diabetes, 55(2), 326-33. [DOI:10.2337/diabetes.55.02.06.db05-0520] [PMID]
Pellow, Sh., Chopin, P., File, S. E., & Briley, M. (1985). Validation of open: Closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. Journal of Neuroscience Methods, 14(3), 149-67. [DOI:10.1016/0165-0270(85)90031-7]
Pilling, A. D., Horiuchi, D., Lively, C. M., & Saxton, W. M. (2006). Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Molecular Biology of the Cell, 17(4), 2057-68. [DOI:10.1091/mbc.e05-06-0526] [PMID] [PMCID]
Pittenger, C., & Duman, R. S. (2008). Stress, depression, and neuroplasticity: A convergence of mechanisms. Neuropsychopharmacology, 33(1), 88-109. [DOI:10.1038/sj.npp.1301574] [PMID]
Prodanov, D., & Feirabend, H. K. P. (2007). Morphometric analysis of the fiber populations of the rat sciatic nerve, its spinal roots, and its major branches. Journal of Comparative Neurology, 503(1), 85-100. [DOI:10.1002/cne.21375] [PMID]
Rajashree, R., Kholkute, S. D., & Goudar, S. S. (2011). Effects of duration of diabetes on behavioural and cognitive parameters in streptozotocin-induced juvenile diabetic rats. The Malaysian Journal of Medical Sciences, 18(4), 26-31. [PMID] [PMCID]
Reske-Nielsen, E., Lundbaek, K., & Rafaelsen, O. J. (1966). Pathological changes in the central and peripheral nervous system of young long-term diabetics: I. diabetic encephalopathy. Diabetologia, 1(3-4), 233-41. [DOI:10.1007/BF01257917] [PMID]
Rivera, E. J., Goldin, A., Fulmer, N., Tavares, R., Wands, J. R., & de la Monte, S. M. (2005). Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. Journal of Alzheimer’s Disease, 8(3), 247-68. [DOI:10.3233/JAD-2005-8304] [PMID]
Roghani, M., Joghataie, M. T., Jalali, M. R., & Baluchnejadmojarad, T. (2006). Time course of changes in passive avoidance and Y-maze performance in male diabetic rats. Iranian Biomedical Journal, 10(2), 99-104. http://ibj.pasteur.ac.ir/article-1-355-en.html
Russo, G. J., Louie, K., Wellington, A., Macleod, G. T., Hu, F., & Panchumarthi, S., et al. (2009). Drosophila Miro is required for both anterograde and retrograde axonal mitochondrial transport. The Journal of Neuroscience, 29(17), 5443-55. [DOI:10.1523/JNEUROSCI.5417-08.2009] [PMID] [PMCID]
Sima, A. A. F., & Li, Z. G. (2005). The effect of C-peptide on cognitive dysfunction and hippocampal apoptosis in type 1 diabetic rats. Diabetes, 54(5), 1497-505. [DOI:10.2337/diabetes.54.5.1497] [PMID]
Sima, A. A. F., Zhang, W., Muzik, O., Kreipke, C. W., Rafols, J. A., & Hoffman, W. H. (2009). Sequential abnormalities in type 1 diabetic encephalopathy and the effects of C-Peptide. The Review of Diabetic Studies, 6(3), 211-22. [DOI:10.1900/RDS.2009.6.211] [PMID] [PMCID]
Steen, E., Terry, B. M., Rivera, E. J., Cannon, J. L., Neely, T. R., & Tavares, R., et al. (2005). Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease -- is this type 3 diabetes? Journal of Alzheimer’s Disease, 7(1), 63-80. [DOI:10.3233/JAD-2005-7107] [PMID]
Stewart, R., & Liolitsa, D. (1999). Type 2 diabetes mellitus, cognitive impairment and dementia. Diabetic Medicine, 16(2), 93-112. [DOI:10.1046/j.1464-5491.1999.00027.x] [PMID]
Strachan, M. W. J. (2011). R D Lawrence Lecture 2010. The brain as a target organ in type 2 diabetes: Exploring the links with cognitive impairment and dementia. Diabetic Medicine, 28(2), 141-7. [DOI:10.1111/j.1464-5491.2010.03199.x] [PMID]
Stranahan, A. M., Arumugam, T. V., Cutler, R. G., Lee, K., Egan, J. M., & Mattson, M. P. (2008). Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nature Neuroscience, 11(3), 309-17. [DOI:10.1038/nn2055] [PMID] [PMCID]
Tang, Z. J., Zou, W., Yuan, J., Zhang, P., Tian, Y., & Xiao, Z. F., et al. (2015). Antidepressant-like and anxiolytic-like effects of hydrogen sulfide in streptozotocin-induced diabetic rats through inhibition of hippocampal oxidative stress. Behavioural Pharmacology, 26(5), 427-35. [DOI:10.1097/FBP.0000000000000143] [PMID]
Wang, A., Lucero, E., Bennett, S., & Potter, H. (2016). O4‐03‐04: Type 3 diabetes in Alzheimer’s disease: Ab‐mediated inhibition of eg5/kinesin 5 leads to insulin receptor and glucose transporter mis‐localization and dysfunction. Alzheimer’s & Dementia, 12(7S Pt 7), 338. [DOI:10.1016/j.jalz.2016.06.621]
Zalsman, G., Huang, Y. Y., Oquendo, M. A., Burke, A. K., Hu, X. Z., & Brent, D. A., et al. (2006). Association of a triallelic serotonin transporter gene promoter region (5-HTTLPR) polymorphism with stressful life events and severity of depression. The American Journal of Psychiatry, 163(9), 1588-93. [DOI:10.1176/ajp.2006.163.9.1588] [PMID]
Zhao, C., Takita, J., Tanaka, Y., Setou, M., Nakagawa, T., & Takeda, S., et al. (2001). Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell, 105(5), 587-97. [DOI:10.1016/S0092-8674(01)00363-4]
Zilliox, L. A., Chadrasekaran, K., Kwan, J. Y., & Russell, J. W. (2016). Diabetes and cognitive impairment. Current Diabetes Reports, 16(9), 87. [DOI:10.1007/s11892-016-0775-x] [PMID] [PMCID]
Full-Text:
1. Introduction
Diabetic encephalopathy and or type 3 Diabetes Mellitus (T3DM) is the chronic complication of Diabetes Mellitus (DM) (Steen et al., 2005; Rivera et al., 2005; de la Monte & Wands, 2008; Leszek, Trypka; & Tarasov, 2017). Any cognitive disorder aligned with DM, in the absence of other predisposing factors, is considered as diabetic encephalopathy or T3DM (Steen et al., 2005, Rivera et al., 2005). Diabetic encephalopathy includes various pathologies and neurobehavioral changes in the nervous system, such as impairment of memory, decision making, and even mood disorders. In many experimental studies, cognitive impairments induced by diabetes or diabetic encephalopathy were observed in different behavioral tests. For example, anxiety-like behavior increased following Type 1 Diabetes Mellitus (T1DM) in rodents (Miyata, Yamada, Hirano, Tanaka, & Kamei, 2007). Also learning and memory deficit were observed in behavioral tests in diabetic animals (Mousavi, Eidi, & Khalili, 2018; Roghani, Joghataie, Jalali, & Baluchnejadmojarad, 2006). In this research, they found a significant cognitive decline in passive avoidance, and Y maze tests diabetic rats.
Hyperglycemic conditions (Reske-Nielsen, Lundbaek, & Rafaelsen, 1966) decreased serum insulin and c-peptide level, cerebrovascular change (Brands, Bell, & Gibson. 2004, Sima, & Li, 2005), neurotropic agent loss (Connor et al., 1997; Lupien, Bluhm, & Ishii, 2003), NF𝜅B pathway and inflammation (Cai & Liu, 2012) and oxidative stress (Beckman & Ames, 1998) are all believed to be responsible for such neurodegenerative condition. In addition to hyperglycemic conditions, low insulin concentration decreased neurotrophic factors, mitochondrial dysfunction is a well-known and confirmed factor to induce T3DM. Mitochondrial dysfunction, on the other hand, is suggested to be the triggering factor for T3DM or Alzheimer disease (Belkacemi & Ramassamy, 2012). Mitochondrial dysfunction is responsible for both acute and chronic hepatic encephalopathy (Bessman & Bessman, 1955; Hindfelt, Plum, & Duffy, 1977). In MELAS (Mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes), which is a genetic condition of the mitochondrial genome, the correlation of mitochondrial dysfunction and encephalopathy have been revealed (Finsterer, 2006). Neural microtubules are arranged polar, with a negative pole in the soma and positive in the axon terminal. Mitochondrial traffic inside the axon is carried out by two important motor proteins: kinesin and dynein. Mitochondrial viability is dependent on its normal transport and movement along the axon via transport proteins (Hollenbeck & Saxton, 2005).
The compromised mitochondrial movement, following axonal transport defect, will impair normal mitochondrial membrane potential, ATP production, and finally result in neurodegeneration (Zhao et al., 2001; Baloh, Schmidt , Pestronk,& Milbrandt, 2007; Guo et al., 2017). Among axonal transport proteins, kinesin and dynein play a key role. For example in fatal encephalopathy, a severe neurodevelopmental disorder, which is associated with hippocampal dysfunction, impaired mitochondrial function is present (Haas et al., 2007), in addition to trafficking kinesin proteins deficiency (Barel et al., 2017). Impaired kinesin and mitochondrial function have been reported in diabetes pathophysiology, as well (Lu et al., 2004; Wang, Bennett, Potter, & Potter, 2016; Zilliox, Chadrasekaran, Kwan, & Russell, 2016). In this study, we used different behavioral tests to prove the cognitive impairments in diabetes and tried to evaluate the correlation of mitochondrial function, kinesin and dynein gene expression following diabetic encephalopathy.
2. Methods
2.1. Animals
Twenty-four male Wistar rats weighing 200-250 g were purchased from the experimental and comparative studies center of Iran University of Medical Sciences, Tehran, Iran. The rats were housed 1 week before the study at room temperature of 22°C±2°C under a 12 h light/dark cycle and they had free access to food and water ad libitum. The animals were housed in a group of 4 rats per cage and were randomly divided into three groups (n= 8 for each group): 1. Control + saline; 2. Diabetic, and 3. Diabetic + insulin.
All of the procedures in use were under the supervision of the Ethics Committee of “Iran University of Medical Sciences”, (Ethical Code: IR.IUMS.REC,1395.9221343204). All procedures are based on ethical guidelines for the care and use of laboratory animals, published by the National Institutes of Health (NIH Publication, revised 1996).
2.2. Diabetes induction
All animals were checked for bodyweight and plasma glucose levels first and rats with Fasting Blood Sugar (FBS) below 150 mg/dL entered the exam. Following overnight fasting, a single dose of intraperitoneal streptozotocin (STZ, Sigma Aldrich, USA), 60 mg/kg dissolved in cold normal saline induced diabetes mellitus (Abebe, Harris, & Macleod, 1990). The volume of the infused solution for each rat was 0.5 mL. The control + saline group received an equivalent volume of saline solution (0.9%) for 8 weeks. The rats with FBS over 150 mg/dL are considered as diabetes. In the diabetes group, there was no treatment at the same time. The diabetic + insulin group was treated with insulin (1.5 U, NPH, 2 times/day) for 8 weeks. Bodyweight and FBS were measured one week after STZ injection and at the end of the eighth week, before behavioral studies.
2.3. Behavioral studies
2.3.1. Elevated Plus Maze (EPM)
After the end of the eighth week, explorative activity and anxiety behavior were assessed using the Elevated Plus Maze (EPM) task. EPM has four arms (plus-shaped), 60 cm in length, 10 cm in width, and 50 cm above the ground. Two arms were enclosed by 30-cm height walls and open arms with 0.5 cm edge. In this experiment, each rat was placed at the junction of the open and closed with the head toward open arms and permitted to seek the arms for 5 min. The total time of presence in both open arms (OAT) and closed arm (CAT) was measured. Decreased presence time in OAT was considered as anxiety-like behavior (Arfa-Fatollahkhani et al., 2017). The number of entries to open arm (%) and time spent in it (%) revealed anxiety indices (Hritcu, Cioanca, & Hancianu, 2012). After each test, the instrument was carefully cleaned with wet tissue (75% ethanol).
2.3.2. Y-maze
About 48 h after the elevated plus-maze task, the animals were subjected to working memory performance by recording spontaneous alternation behavior in the Y-maze task. The maze was made of a Y-shaped Plexiglas holding consists of three arms (A, B, C). The arms are converged in an equilateral triangular (1200) with 40-cm length, 30-cm height, and 15-cm width. Each rat was placed at the beginning of one arm “A” and permitted to discover the apparatus for 8 min. An entry occurred when all four limbs were inside the arm. Spontaneous alternation was defined as successive entries into the three arms on triplet sets. The spontaneous alternation percentage was calculated as the ratio of actual to possible alternations (defined as the total number of arm entries minus two multiply by 100) (Roghani, Joghataie & Jalali, 2006; Mousavi et al., 2018). After each test, the instrument was carefully cleaned with wet tissue (75% ethanol).
2.3.3. Passive avoidance learning
About 48 hours after Y-maze, passive avoidance learning (PAL) was performed in a shuttle box. The shuttle box was used to evaluate passive avoidance behavior. The apparatus consists of light and dark compartments of equal size (20×40×20 cm) connected by a small central guillotine door. Electric shock was received by grid floor, in the dark compartment. This test was performed within 4 days. In the first and second days, all rats were adapted to the apparatus for 5 min. On the third day, each rat settled in the light compartment for 2 min, then the guillotine door was opened then the latency time to enter the dark compartment was recorded as initial latency (IL). When the rats entered the dark compartment, the guillotine door was closed and electric foot shock (1 s, 1 mA, 50 Hz) was received. The rats with IL higher than 60 s were excluded. To evaluate memory retention, each rat was placed again in the light compartment the next day. The interval between the entrance to the light chamber and leaving into the dark chamber was measured as step-through latency (STL). The STL was measured as the index for passive avoidance behavior (cut-off time was 480 ) . (Roghani et al., 2006) After each test, the apparatus was carefully cleaned with wet tissue (75% ethanol).
2.3.4. Gene expression of KIF5b and Dynein by real-time PCR
After behavioral tests, the animals were deeply and irreversibly anesthetized, then the rats were sacrificed, the skull was opened, the brain was removed and washed with normal saline, hemispheres of the brain were separated by a scalpel blade and the midbrain was removed. Then, the hippocampus was dissected and tissue was stored at -80°C. KIF5b and dynein gene expression were determined using real-time polymerase chain reactions (R-T PCR). The total RNA was extracted using RNX-plus solution (SinaClon, Iran), 75% ethanol, chloroform, diethyl pyrocarbonate (DEPC)-treated water, and isopropanol following the manufacturer’s recommendations. The concentration of RNA was checked by a UV spectrophotometer (Ultrospec 2000, Pharmacia, the Netherlands). By measuring Optical Density (OD) at a wavelength A260/A280 nm, the quantity of the isolated RNA was determined. The first strand cDNA was generated from 1µg of total RNA by reverse transcription using PrimScript RT reagent Kit (Takara, Japan), based on the manufacturer’s instructions and R-T PCR assays were down in 72-well plates in a Rotor-Gene 6000 device (Corbett Life Science, Australia).
The forward and reverse primers for dynein gene:
Forward primer: TGCTTGGAAGATGATTGTGC
Reverse primer: TTCTCTTCCTCGGTCAACTCA
The forward and reverse primers for the KIF5b gene:
Forward primer: TGCCTATTGATGAGCAGTTTG
Reverse primer: GCCGGTTTGTCATTGGTAAT
The mRNA expression of KIF5b, dynein and ß-actin (as housekeeping) were determined. The PCR volume reaction including 1 µL cDNA, 5µL SYBER Premix Ex Taq (Takara, Japan), 0.5 µL forward primer, and 0.5 µL reverse primer was 10 µL.
The analysis of real-time PCR was performed with ∆∆Ct method. The fold change in expression was then obtained as 2-∆∆CT (Prodanov & Feirabend, 2007).
2.3.5. Measurement of mitochondrial membrane potential by rhodamine 123 probe
Mitochondrial membrane potential (MMP) was assessed by monitoring the uptake level of cationic dye rhodamine 123 (Mingatto et al., 2002). Hippocampus containing blocks were prepared, washed with phosphate-buffered saline (PBS), and homogenized in mitochondrial isolation buffer (0.01 mol/L Tris HCL, 0.0001 mol/L EDTA 2Na [ethylenediaminetetracetic acid disodium], 0.01 mol/L sucrose, 0.8% NaCl, pH=7.4) on ice (00C) for about 2 min. The homogenate was kept at 40C and centrifuged at 1500 rpm for 10 min. The supernatant was collected and then centrifuged again at 10000 rpm for 15 min at 40C. The remaining part was mitochondrial. Then, 20 µL of rhodamine 123 solution (rhodamine 123 solution 1 mg/10 mL dimethyl sulfoxide) and 180 µL of PBS was added to it, stirred, transferred to 96-well plate, and incubated at 37°C for 30 min. Afterward, the MMP was studied. Fluorescent signals of mitochondria were cleared at 488 nm and emission was monitored at 525 nm wavelength in a fluorescent plate reader (FLX 800, BioTek, USA) (Ding et al., 2013).
2.3.6. Blood sampling and biochemical parameters
After behavioral tests, the rats were sacrificed and their blood samples were taken. To measure the serum level of insulin, blood samples were kept at room temperature for 2 h to clot, then they were centrifuged at 5000 rpm for 20 min (Lulat, Yadav, Balaraman, & Maheshwari, 2016). The serum was stored at -20°C. Repeated freezing and defrosting were avoided. To measure glycosylated hemoglobin (HgbA1c), the whole blood was collected. Serum levels of insulin and HgbA1c were measured using ELISA kits, according to the manufacturer instructions for insulin (Mercodia, Sweden) and HgbA1c (Roch, Germany).
2.3.7. Statistical analysis
Data analysis was performed with GraphPad Prism 7 software. All data are represented as Mean±SEM. The obtained data were assessed by 1-way analysis of variance (ANOVA) followed by Tukey post-hoc test except for body weight and FBS. To compare the results of body weight and FBS, 2-way repeated measures (ANOVA) followed by Bonferroni post-hoc test were used. The value of P<0.05 was considered statistically significant.
3. Results
3.1. Bodyweight, fasting blood sugar
Before starting the experiments, all animals were checked for blood glucose levels and animals with levels less than 150 mg/dL entered the exam. Bodyweight and FBS were measured at the first and eighth weeks after STZ injection. The Mean±SEM weight and FBS of the rats are presented in Table 1 The bodyweight of animals in groups was similar in the first week.
Diabetic encephalopathy and or type 3 Diabetes Mellitus (T3DM) is the chronic complication of Diabetes Mellitus (DM) (Steen et al., 2005; Rivera et al., 2005; de la Monte & Wands, 2008; Leszek, Trypka; & Tarasov, 2017). Any cognitive disorder aligned with DM, in the absence of other predisposing factors, is considered as diabetic encephalopathy or T3DM (Steen et al., 2005, Rivera et al., 2005). Diabetic encephalopathy includes various pathologies and neurobehavioral changes in the nervous system, such as impairment of memory, decision making, and even mood disorders. In many experimental studies, cognitive impairments induced by diabetes or diabetic encephalopathy were observed in different behavioral tests. For example, anxiety-like behavior increased following Type 1 Diabetes Mellitus (T1DM) in rodents (Miyata, Yamada, Hirano, Tanaka, & Kamei, 2007). Also learning and memory deficit were observed in behavioral tests in diabetic animals (Mousavi, Eidi, & Khalili, 2018; Roghani, Joghataie, Jalali, & Baluchnejadmojarad, 2006). In this research, they found a significant cognitive decline in passive avoidance, and Y maze tests diabetic rats.
Hyperglycemic conditions (Reske-Nielsen, Lundbaek, & Rafaelsen, 1966) decreased serum insulin and c-peptide level, cerebrovascular change (Brands, Bell, & Gibson. 2004, Sima, & Li, 2005), neurotropic agent loss (Connor et al., 1997; Lupien, Bluhm, & Ishii, 2003), NF𝜅B pathway and inflammation (Cai & Liu, 2012) and oxidative stress (Beckman & Ames, 1998) are all believed to be responsible for such neurodegenerative condition. In addition to hyperglycemic conditions, low insulin concentration decreased neurotrophic factors, mitochondrial dysfunction is a well-known and confirmed factor to induce T3DM. Mitochondrial dysfunction, on the other hand, is suggested to be the triggering factor for T3DM or Alzheimer disease (Belkacemi & Ramassamy, 2012). Mitochondrial dysfunction is responsible for both acute and chronic hepatic encephalopathy (Bessman & Bessman, 1955; Hindfelt, Plum, & Duffy, 1977). In MELAS (Mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes), which is a genetic condition of the mitochondrial genome, the correlation of mitochondrial dysfunction and encephalopathy have been revealed (Finsterer, 2006). Neural microtubules are arranged polar, with a negative pole in the soma and positive in the axon terminal. Mitochondrial traffic inside the axon is carried out by two important motor proteins: kinesin and dynein. Mitochondrial viability is dependent on its normal transport and movement along the axon via transport proteins (Hollenbeck & Saxton, 2005).
The compromised mitochondrial movement, following axonal transport defect, will impair normal mitochondrial membrane potential, ATP production, and finally result in neurodegeneration (Zhao et al., 2001; Baloh, Schmidt , Pestronk,& Milbrandt, 2007; Guo et al., 2017). Among axonal transport proteins, kinesin and dynein play a key role. For example in fatal encephalopathy, a severe neurodevelopmental disorder, which is associated with hippocampal dysfunction, impaired mitochondrial function is present (Haas et al., 2007), in addition to trafficking kinesin proteins deficiency (Barel et al., 2017). Impaired kinesin and mitochondrial function have been reported in diabetes pathophysiology, as well (Lu et al., 2004; Wang, Bennett, Potter, & Potter, 2016; Zilliox, Chadrasekaran, Kwan, & Russell, 2016). In this study, we used different behavioral tests to prove the cognitive impairments in diabetes and tried to evaluate the correlation of mitochondrial function, kinesin and dynein gene expression following diabetic encephalopathy.
2. Methods
2.1. Animals
Twenty-four male Wistar rats weighing 200-250 g were purchased from the experimental and comparative studies center of Iran University of Medical Sciences, Tehran, Iran. The rats were housed 1 week before the study at room temperature of 22°C±2°C under a 12 h light/dark cycle and they had free access to food and water ad libitum. The animals were housed in a group of 4 rats per cage and were randomly divided into three groups (n= 8 for each group): 1. Control + saline; 2. Diabetic, and 3. Diabetic + insulin.
All of the procedures in use were under the supervision of the Ethics Committee of “Iran University of Medical Sciences”, (Ethical Code: IR.IUMS.REC,1395.9221343204). All procedures are based on ethical guidelines for the care and use of laboratory animals, published by the National Institutes of Health (NIH Publication, revised 1996).
2.2. Diabetes induction
All animals were checked for bodyweight and plasma glucose levels first and rats with Fasting Blood Sugar (FBS) below 150 mg/dL entered the exam. Following overnight fasting, a single dose of intraperitoneal streptozotocin (STZ, Sigma Aldrich, USA), 60 mg/kg dissolved in cold normal saline induced diabetes mellitus (Abebe, Harris, & Macleod, 1990). The volume of the infused solution for each rat was 0.5 mL. The control + saline group received an equivalent volume of saline solution (0.9%) for 8 weeks. The rats with FBS over 150 mg/dL are considered as diabetes. In the diabetes group, there was no treatment at the same time. The diabetic + insulin group was treated with insulin (1.5 U, NPH, 2 times/day) for 8 weeks. Bodyweight and FBS were measured one week after STZ injection and at the end of the eighth week, before behavioral studies.
2.3. Behavioral studies
2.3.1. Elevated Plus Maze (EPM)
After the end of the eighth week, explorative activity and anxiety behavior were assessed using the Elevated Plus Maze (EPM) task. EPM has four arms (plus-shaped), 60 cm in length, 10 cm in width, and 50 cm above the ground. Two arms were enclosed by 30-cm height walls and open arms with 0.5 cm edge. In this experiment, each rat was placed at the junction of the open and closed with the head toward open arms and permitted to seek the arms for 5 min. The total time of presence in both open arms (OAT) and closed arm (CAT) was measured. Decreased presence time in OAT was considered as anxiety-like behavior (Arfa-Fatollahkhani et al., 2017). The number of entries to open arm (%) and time spent in it (%) revealed anxiety indices (Hritcu, Cioanca, & Hancianu, 2012). After each test, the instrument was carefully cleaned with wet tissue (75% ethanol).
2.3.2. Y-maze
About 48 h after the elevated plus-maze task, the animals were subjected to working memory performance by recording spontaneous alternation behavior in the Y-maze task. The maze was made of a Y-shaped Plexiglas holding consists of three arms (A, B, C). The arms are converged in an equilateral triangular (1200) with 40-cm length, 30-cm height, and 15-cm width. Each rat was placed at the beginning of one arm “A” and permitted to discover the apparatus for 8 min. An entry occurred when all four limbs were inside the arm. Spontaneous alternation was defined as successive entries into the three arms on triplet sets. The spontaneous alternation percentage was calculated as the ratio of actual to possible alternations (defined as the total number of arm entries minus two multiply by 100) (Roghani, Joghataie & Jalali, 2006; Mousavi et al., 2018). After each test, the instrument was carefully cleaned with wet tissue (75% ethanol).
2.3.3. Passive avoidance learning
About 48 hours after Y-maze, passive avoidance learning (PAL) was performed in a shuttle box. The shuttle box was used to evaluate passive avoidance behavior. The apparatus consists of light and dark compartments of equal size (20×40×20 cm) connected by a small central guillotine door. Electric shock was received by grid floor, in the dark compartment. This test was performed within 4 days. In the first and second days, all rats were adapted to the apparatus for 5 min. On the third day, each rat settled in the light compartment for 2 min, then the guillotine door was opened then the latency time to enter the dark compartment was recorded as initial latency (IL). When the rats entered the dark compartment, the guillotine door was closed and electric foot shock (1 s, 1 mA, 50 Hz) was received. The rats with IL higher than 60 s were excluded. To evaluate memory retention, each rat was placed again in the light compartment the next day. The interval between the entrance to the light chamber and leaving into the dark chamber was measured as step-through latency (STL). The STL was measured as the index for passive avoidance behavior (cut-off time was 480 ) . (Roghani et al., 2006) After each test, the apparatus was carefully cleaned with wet tissue (75% ethanol).
2.3.4. Gene expression of KIF5b and Dynein by real-time PCR
After behavioral tests, the animals were deeply and irreversibly anesthetized, then the rats were sacrificed, the skull was opened, the brain was removed and washed with normal saline, hemispheres of the brain were separated by a scalpel blade and the midbrain was removed. Then, the hippocampus was dissected and tissue was stored at -80°C. KIF5b and dynein gene expression were determined using real-time polymerase chain reactions (R-T PCR). The total RNA was extracted using RNX-plus solution (SinaClon, Iran), 75% ethanol, chloroform, diethyl pyrocarbonate (DEPC)-treated water, and isopropanol following the manufacturer’s recommendations. The concentration of RNA was checked by a UV spectrophotometer (Ultrospec 2000, Pharmacia, the Netherlands). By measuring Optical Density (OD) at a wavelength A260/A280 nm, the quantity of the isolated RNA was determined. The first strand cDNA was generated from 1µg of total RNA by reverse transcription using PrimScript RT reagent Kit (Takara, Japan), based on the manufacturer’s instructions and R-T PCR assays were down in 72-well plates in a Rotor-Gene 6000 device (Corbett Life Science, Australia).
The forward and reverse primers for dynein gene:
Forward primer: TGCTTGGAAGATGATTGTGC
Reverse primer: TTCTCTTCCTCGGTCAACTCA
The forward and reverse primers for the KIF5b gene:
Forward primer: TGCCTATTGATGAGCAGTTTG
Reverse primer: GCCGGTTTGTCATTGGTAAT
The mRNA expression of KIF5b, dynein and ß-actin (as housekeeping) were determined. The PCR volume reaction including 1 µL cDNA, 5µL SYBER Premix Ex Taq (Takara, Japan), 0.5 µL forward primer, and 0.5 µL reverse primer was 10 µL.
The analysis of real-time PCR was performed with ∆∆Ct method. The fold change in expression was then obtained as 2-∆∆CT (Prodanov & Feirabend, 2007).
2.3.5. Measurement of mitochondrial membrane potential by rhodamine 123 probe
Mitochondrial membrane potential (MMP) was assessed by monitoring the uptake level of cationic dye rhodamine 123 (Mingatto et al., 2002). Hippocampus containing blocks were prepared, washed with phosphate-buffered saline (PBS), and homogenized in mitochondrial isolation buffer (0.01 mol/L Tris HCL, 0.0001 mol/L EDTA 2Na [ethylenediaminetetracetic acid disodium], 0.01 mol/L sucrose, 0.8% NaCl, pH=7.4) on ice (00C) for about 2 min. The homogenate was kept at 40C and centrifuged at 1500 rpm for 10 min. The supernatant was collected and then centrifuged again at 10000 rpm for 15 min at 40C. The remaining part was mitochondrial. Then, 20 µL of rhodamine 123 solution (rhodamine 123 solution 1 mg/10 mL dimethyl sulfoxide) and 180 µL of PBS was added to it, stirred, transferred to 96-well plate, and incubated at 37°C for 30 min. Afterward, the MMP was studied. Fluorescent signals of mitochondria were cleared at 488 nm and emission was monitored at 525 nm wavelength in a fluorescent plate reader (FLX 800, BioTek, USA) (Ding et al., 2013).
2.3.6. Blood sampling and biochemical parameters
After behavioral tests, the rats were sacrificed and their blood samples were taken. To measure the serum level of insulin, blood samples were kept at room temperature for 2 h to clot, then they were centrifuged at 5000 rpm for 20 min (Lulat, Yadav, Balaraman, & Maheshwari, 2016). The serum was stored at -20°C. Repeated freezing and defrosting were avoided. To measure glycosylated hemoglobin (HgbA1c), the whole blood was collected. Serum levels of insulin and HgbA1c were measured using ELISA kits, according to the manufacturer instructions for insulin (Mercodia, Sweden) and HgbA1c (Roch, Germany).
2.3.7. Statistical analysis
Data analysis was performed with GraphPad Prism 7 software. All data are represented as Mean±SEM. The obtained data were assessed by 1-way analysis of variance (ANOVA) followed by Tukey post-hoc test except for body weight and FBS. To compare the results of body weight and FBS, 2-way repeated measures (ANOVA) followed by Bonferroni post-hoc test were used. The value of P<0.05 was considered statistically significant.
3. Results
3.1. Bodyweight, fasting blood sugar
Before starting the experiments, all animals were checked for blood glucose levels and animals with levels less than 150 mg/dL entered the exam. Bodyweight and FBS were measured at the first and eighth weeks after STZ injection. The Mean±SEM weight and FBS of the rats are presented in Table 1 The bodyweight of animals in groups was similar in the first week.
Significant impairment in weight gain occurred in the diabetic rats comparing to the control rats in the eighth week after the induction of diabetes (F2,21=151.06, P=0.0001). Also in the diabetic group, FBS significantly increased compared to the control group (F2,21=305.57, P=0.0001). Insulin injection for 8 weeks improved weight gain in diabetic rats. Animals’ weight in the diabetic + insulin was significantly higher than that in the diabetic rats (P<0.05). FBS was significantly lower than that in the diabetic rats (P<0.05) (Table 1).
3.2. Behavioral tests
3.2.1. Elevated Plus Maze (EPM)
The number of entries to open arm and time spent in open arms were considered as anxiety indices and presented as a percentage. Time spent in open arms (%) was significantly decreased (F2,18=4.88, P=0.02) (Figure 1).
This index decreased in diabetic rat compared to that in the control group (control + saline = 61.88±20.14, diabetic=21.18±18.49, P<0.05). Whereas the number of open arm entrance (%) was not changed in groups (F2,18=2.26, P=0.1324) (Figure 2).
3.2.2. Y-maze
Y-maze test was performed 48 hours after EPM. The percentage of alternation behavior 8 weeks after the induction of diabetes was significantly changed (F2,18=3.60, P=0.048) (Figure 3).
This index decreased in diabetic rats compared to the control group (control + saline =28.57±5.53, diabetic=15.57±11.77, P<0.05).
3.2.3. Passive Avoidance Learning (PAL)
This test was performed 48 h after the Y-maze task. The initial latency was recorded when the rats entered the dark compartment. In all three groups, there was no significant change in IL (F2,18=0.686, P=0.515) (Figure 4).
The step-through latency (maximum 480 s) was measured and recorded as the index for passive avoidance learning. STL index was significantly changed (F2,18=7.31, P=0.0047) (Figure 5).
STL in the diabetic group decreased compared to that in the control group (control + saline =384±123.94, diabetic=95.28±70.77, P<0.05).
3.2.4. Gene expression of KIF5b and Dynein by real-time PCR
Real-time PCR was carried out to reveal KIF5b and dynein gene expression (Figure 6).
Gene expression of KIF5b dramatically changed after 8 weeks of diabetes induction (F2,9=20.41, P=0.0005). Actually, KIF5b mRNA levels were significantly increased compared to the control group (control + saline = 1±0.4, diabetic = 2.25±0.47, P<0.05). Insulin injection for 8 weeks significantly decreased expression of KIF5b compared with diabetic rats (diabetic = 2.25±0.4796, diabetic + insulin=0.5±0.23, P<0.05) (Figure 6). No significant changes were observed in dynein mRNA levels in different groups (F2,9=1.86, P =0.2096) (Figure 7).
3.2.5. Mitochondrial membrane potential by rhodamine 123 probe
The Mitochondrial Membrane Potential (MMP) of cells was assessed by monitoring the uptake of cationic dye rhodamine 123. The results of MMP showed a significant change in different groups (F2,18= 8.557, P=0.0024) (Figure 8).
In the diabetic group, MMP decreased 8 weeks after induction of diabetes compared to that in the control group (control + saline=100.1±12.14, diabetic = 68.05±20.64, P<0.05) and there was a significant difference between the control group and insulin injected groups (control + saline=100.1±12.14, diabetic + insulin=75.88±10.58, P<0.05).
3.2.6. Biochemical parameters
Serum level of insulin and the percentage of glycosylated hemoglobin (HgbA1c %) were measured by using ELISA kits. Mean±SEM of HgbA1c (%) and serum insulin are given in Table 2 Results of HgbA1c percentage showed a significant difference between the groups (F2,18=95.99, P=0.0001).
Diabetic animals revealed higher glycosylated hemoglobin compared to age-matched controls (P<0.05). A significant difference was observed between the diabetic and insulin-treated group (P<0.05).
The serum level of insulin was significantly different between the groups (F2,12= 316.3, P=0.0001). Insulin levels in diabetic rats were significantly lower than control rats (P<0.05). This difference was cleared between the control and insulin-treated rats (P<0.05) as well.
4. Discussion
Diabetic encephalopathy is referred to as any cognitive dysfunction following diabetes mellitus. This is why this condition is called type 3 DM (T3DM) or Alzheimer disease (Steen et al., 2005; Rivera et al., 2005; de la Monte, & Wands, 2008; Leszek et al., 2017). On the other hand, Mitochondrial Diabetes (MD) appears after the activation of mutant mitochondrial DNA, which is age-dependent. MD is the combination of diabetes and cognitive deficit (Maassen et al., 2004; Maassen, Janssen, & Hart, 2005). In this research, we showed the correlation between diabetic encephalopathy, dysfunctional mitochondria, and change in the expression of axonal transport protein kinesin (KIF5b).
We showed short-term memory impairment and retrieval deficit in combination with anxiety-like behavior following T3DM. To measure anxiety-like behavior in rodents, the elevated plus maze is an accepted test (Pellow, Chopin, File, & Briley, 1985). Our findings on EPM showed a decrease in time spent in open arm, in the diabetic group, compared with the control and or insulin-injected groups. In 2007, Miyata showed an increase of anxiety-like behavior following STZ-induced T1DM in rodents (Miyata et al., 2007).
Another study has revealed that N-acetylcysteine (NAC) is protective against anxiety-like behavior in the EPM test of diabetic rats (Kamboj, Chopra, & Sandhir, 2008). Behavioral changes due to diabetic encephalopathy are not only dependent on the insulin metabolism cascade impairment, but also because of decreased serotonin levels in the central nervous system (Zalsman et al., 2006, Pittenger & Duman, 2008). On the other hand, hyperglycemia induces neurodegeneration and pathologic behavior and cognition. In the diabetic + insulin group, insulin has possibly prevented behavioral deficits. Our findings in the passive avoidance learning task showed different degrees of learning and memory impairment in type 3 diabetes. Actually, in the shuttle box, the significant decrease in Step-Through Latency (STL) in T3DM is referred to impairment of memory retention and consolidation. Scientists have reported the role of insulin on the improvement of memory in DM, which is in agreement with our results (diabetic + insulin group showing an increase in STL by insulin administration). Following prolonged DM, the impaired hippocampal plasticity may damage learning and memory processing (Flood, Mooradian, & Morley, 1990; Stewart & Liolitsa, 1999; Stranahan et al., 2008). Scientists have found that anxiety level and cognitive dysfunction is directly correlated to diabetes duration in young rats (Rajashree, Kholkute, & Goudar, 2011). Memory and learning deficit and even behavioral changes are among obvious manifestations of diabetic encephalopathy, as insulin and c-peptide are key factors for synaptic plasticity and cognition (Sima et al., 2009; McNay & Recknagel, 2011). Scientists have revealed the role of ICV streptozotocin in the induction of cognitive dysfunction, which has been reversed after insulin infusion (Guo et al., 2017). Physiologic mitochondrial function is dependent on its intact transport process (Chada & Hollenbeck, 2004; Russo et al., 2009). Low insulin and c-peptide levels may disrupt normal cerebral metabolism and function sequentially (de la Monte & Wands, 2008; Sima et al., 2009).
Although our finding was in favor of the late course of T1DM or T3DM, the hypoglycemic course is related to cerebral malfunction is not clear. However, the chronicity of T1DM is correlated with encephalopathy (Garg, Bonanome, Grundy, Zhang, & Unger, 1988; Musen et al., 2006).
Natural flavonoids have been effective in the reversal of anxiety-like behavior detected in the EPM test in diabetic mice (Damian et al., 2014; Tang et al., 2015). Insulin plays a critical role in hippocampal memory processing and hyperglycemia is responsible for the cognitive deficit and neuronal damage following DM (Abbatecola et al., 2006; Strachan et al., 2011).
In rhodamine monitoring of hippocampal mitochondrial membrane potential, we found a decreased level of mitochondrial staining. Mitochondrial function can be monitored by MMP (Figure 9).
The normal detectable electrochemical gradient of mitochondrial membrane refers to ATP production (Hafner, Brown, & Brand, 1990; Fontaine, Devin, Rigoulet, & Leverve, 1997). Rhodamine 123 transmembrane distribution reveals membrane potential strength (Emaus, Grunwald, & Lemasters, 1986; Huang, Camara, Stowe, Qi, & Beard, 2007). Following any mitochondrial and cellular damage, there will be abnormal or undetectable MMP and ATP production and neuronal apoptosis (Letai 2006; de la Monte, 2012). Even low MMP represents apoptosis (Ding, Han, Zhu, Chen, & D’Ambrosio, 2005). Electrophoretic mobility of mitochondria is directly related to its permeability and staining specifies (Emauset al., 1986; Bunting, 1992). In 2017, scientists have shown that normal mitochondrial metabolism potentially prevents from decreased age-dependent neurogenesis in the hippocampus (Beckervordersandforth et al., 2017). Besides mitochondrial count and morphology, mitochondrial metabolism also depends on the dynamic movement and distribution of these organelles, which is essential to maintain normal ATP synthesis (Lu et al., 2004).
Our study showed a higher expression of KIF5b, a member of the kinesin superfamily. KIF5b has a key role in both mitochondrial translocation and distribution. KIF5b depletion will stop normal mitochondrial transport. In 2011 and for the first time, scientists showed the vital role of kinesin protein in mitochondrial transport of hippocampal neurons (Brickley & Anne Stephenson, 2011).
In 2013, scientists demonstrsted increased KIF5b gene expression in cultured hippocampal neurons of diabetic rats with no alterations in dynein gene expression (Baptista, Pinto, Elvas, Almeida, & Ambrósio, 2013). Dynein is not only responsible for retrograde translocation of mitochondria, but responsible for the movement of other motor adaptors (King & Schroer, 2000, Pilling, Horiuchi, Lively, & Saxton, 2006). Although some kinesins are also involved in retrograde transport (Hirokawa, Noda, Tanaka, & Niwa, 2009).
In this study, we found that diabetes alters the KIF5b motor protein by increasing KIF5b gene expression levels in the hippocampus 8 weeks after the induction of diabetes and the anterograde transport of mitochondria may be impaired in the hippocampus. As a consequence, we suggested that a decrease in the number of synaptic vesicles and density may ultimately account for changes in synaptic transmission and mitochondrial transport in the hippocampus. In 2014, the effect of T1DM (duration of 2 to 8 weeks) on rat retinal distribution of dynein, KIF5b, and KIF1a in retinal tissue was confirmed (Baptista et al., 2014), which was considered as the possible factor for diabetic retinal neurodegeneration.
In this study, we found no change in dynein heavy chain expression. Although to our knowledge there is still no report about encephalopathy and dynein gene expression, it may be a more chronic type of T3DM model (longer than 8 weeks) that could reveal different findings. While IHC, as a qualitative method and western blotting as a quantitative one, shows protein end product and complete study, when mRNA production is impaired no protein will be found.
In conclusion, this study revealed the correlation between diabetic encephalopathy, mitochondrial dysfunction, and kinesin gene overexpression. We suggest that abnormal translocation of mitochondria and its malfunction induced by kinesin gene overexpression may result in diabetic encephalopathy.
Ethical Considerations
Compliance with ethical guidelines
All ethical principles are considered in this article in accordance with Physiology Research Center and Cellular and Molecular Research Center of Iran University of Medical Sciences.
Funding
This research did not receive any specific grant or budget from funding agencies in the public, commercial, or not-for-profit sectors.
Authors' contributions
All authors equally contributed to preparing this article.
Conflict of interest
The authors declared no conflict of interest to the publication of this article.
Acknowledgments
We would like to thank our colleagues from the Physiology Research Center and Cellular and Molecular Research Center of Iran University of Medical Sciences.
References
Abbatecola, A. M., Rizzo, M. R., Barbieri, M., Grella, R., Arciello, A., & Laieta, M. T., et al. (2006). Postprandial plasma glucose excursions and cognitive functioning in aged type 2 diabetics. Neurology, 67(2), 235-40. [DOI:10.1212/01.wnl.0000224760.22802.e8] [PMID]
Abebe, W., Harris, K. H., & MacLeod, K. M. (1990). Enhanced contractile responses of arteries from diabetic rats to alpha 1-adrenoceptor stimulation in the absence and presence of extracellular calcium. Journal of Cardiovascular Pharmacology, 16(2), 239-48. [DOI:10.1097/00005344-199008000-00010] [PMID]
Arfa-Fatollahkhani, P., Nahavandi, A., Abtahi, H., Anjidani, S., Borhani, S., & Jameie, S. B., et al. (2017). The effect of luteinizing hormone reducing agent on anxiety and novel object recognition memory in gonadectomized rats. Basic and Clinical Neuroscience, 8(2), 113-9. [DOI:10.18869/nirp.bcn.8.2.113] [PMID] [PMCID]
Baloh, R. H., Schmidt, R. E., Pestronk, A., & Milbrandt, J. (2007). Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. The Journal of Neuroscience, 27(2), 422-30. [DOI:10.1523/JNEUROSCI.4798-06.2007] [PMID] [PMCID]
Baptista, F. I., Pinto, M. J., Elvas, F., Almeida R. D., & Ambrósio, A. F. (2013). Diabetes alters KIF1A and KIF5B motor proteins in the hippocampus. PLoS One, 8(6), e65515. [DOI:10.1371/journal.pone.0065515] [PMID] [PMCID]
Baptista, F. I., Pinto, M. J., Elvas, F., Martins, T., Almeida, R. D., & Ambrosio, A. F. (2014). Diabetes induces changes in KIF1A, KIF5B and dynein distribution in the rat retina: Implications for axonal transport. Experimental Eye Research, 127, 91-103. [DOI:10.1016/j.exer.2014.07.011] [PMID]
Barel, O., Malicdan, M. C. V., Ben-Zeev, B., Kandel, J., Pri-Chen, H., & Stephen, J., et al. (2017). Deleterious variants in TRAK1 disrupt mitochondrial movement and cause fatal encephalopathy. Brain, 140(3), 568-81. [DOI:10.1093/brain/awx002] [PMID] [PMCID]
Beckervordersandforth, R., Ebert, B., Schaffner, I., Moss, J., Fiebig, C., & Shin, J., et al. (2017). Role of mitochondrial metabolism in the control of early lineage progression and aging phenotypes in adult hippocampal neurogenesis. Neuron, 93(6), 1518. [DOI:10.1016/j.neuron.2017.03.008] [PMID] [PMCID]
Beckman, K. B., & Ames, B. N. (1998). The free radical theory of aging matures. Physiological Reviews, 78(2), 547-81. [DOI:10.1152/physrev.1998.78.2.547] [PMID]
Belkacemi, A., & Ramassamy, C. (2012). Time sequence of oxidative stress in the brain from transgenic mouse models of Alzheimer’s disease related to the amyloid-beta cascade. Free Radical Biology and Medicine, 52(3), 593-600. [DOI:10.1016/j.freeradbiomed.2011.11.020] [PMID]
Bessman, S. P., & Bessman, A. N. (1955). The cerebral and peripheral uptake of ammonia in liver disease with an hypothesis for the mechanism of hepatic coma. The Journal of Clinical Investigation, 34(4), 622-8. [DOI:10.1172/JCI103111] [PMID] [PMCID]
Brands, M. W., Bell, T. D., & Gibson, B. (2004). Nitric oxide may prevent hypertension early in diabetes by counteracting renal actions of superoxide. Hypertension, 43(1), 57-63. [DOI:10.1161/01.HYP.0000104524.25807.EE] [PMID]
Brickley, K., Anne Stephenson, F. (2011). Trafficking kinesin protein (TRAK)-mediated transport of mitochondria in axons of hippocampal neuron. The Journal of Biological Chemistry, 286(20), 18079-92. [DOI:10.1074/jbc.M111.236018] [PMID] [PMCID]
Bunting, J. R. (1992). Influx and efflux kinetics of cationic dye binding to respiring mitochondria. Biophysical Chemistry, 42(2), 163-75. [DOI:10.1016/0301-4622(92)85006-P]
Cai, D., & Liu, T. (2012). Inflammatory cause of metabolic syndrome via brain stress and NF-kappaB. Aging, 4(2), 98-115. [DOI:10.18632/aging.100431] [PMID] [PMCID]
Chada, S. R., & Hollenbeck, P. J. (2004). Nerve growth factor signaling regulates motility and docking of axonal mitochondria. Current Biology, 14(14), 1272-6. [DOI:10.1016/j.cub.2004.07.027] [PMID]
Connor, B., Young, D., Yan, Q., Faull, R. L. M., Synek, B., & Dragunow, M. (1997). Brain-derived neurotrophic factor is reduced in Alzheimer’s disease. Molecular Brain Research, 49(1-2), 71-81. [DOI:10.1016/S0169-328X(97)00125-3]
Damian, J. P., Acosta, V., Da Cuña, M., Ramirez, I., Oddone, N., & Zambrana, A., et al. (2014). Effect of resveratrol on behavioral performance of streptozotocin-induced diabetic mice in anxiety tests. Experimental Animals, 63(3), 277-87. [DOI:10.1538/expanim.63.277] [PMID] [PMCID]
de la Monte, S. M. (2012). Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Current Alzheimer Research, 9(1), 35-66. [DOI:10.2174/156720512799015037] [PMID] [PMCID]
de la Monte, S. M., & Wands, J. R. (2008). Alzheimer’s disease is type 3 diabetes-evidence reviewed. Journal of Diabetes Science and Technology, 2(6), 1101-13. [DOI:10.1177/193229680800200619] [PMID] [PMCID]
Ding, H., Han, C., Zhu, J., Chen, C. S., & D’Ambrosio, S. M. (2005). Celecoxib derivatives induce apoptosis via the disruption of mitochondrial membrane potential and activation of caspase 9. International Journal of Cancer, 113(5), 803-10. [DOI:10.1002/ijc.20639] [PMID]
Ding, J., Yu, H. L., Ma, W. W., Xi, Y. D., Zhao, X., & Yuan, L. H., et al. (2013). Soy isoflavone attenuates brain mitochondrial oxidative stress induced by beta-amyloid peptides 1-42 injection in lateral cerebral ventricle. Journal of Neuroscience Research, 91(4), 562-7. [DOI:10.1002/jnr.23163] [PMID]
Emaus, R. K., Grunwald, R., & Lemasters, J. J. (1986). Rhodamine 123 as a probe of transmembrane potential in isolated rat-liver mitochondria: Spectral and metabolic properties. Biochimica et Biophysica Acta (BBA) - Bioenergetics, 850(3), 436-48. [DOI:10.1016/0005-2728(86)90112-X]
Finsterer, J. (2006). Central nervous system manifestations of mitochondrial disorders. Acta neurologica Scandinavica, 114(4), 217-38. [DOI:10.1111/j.1600-0404.2006.00671.x] [PMID]
Flood, J. F., Mooradian, A. D., & Morley, J. E. (1990). Characteristics of learning and memory in streptozocin-induced diabetic mice. Diabetes, 39(11), 1391-8. [DOI:10.2337/diab.39.11.1391] [PMID]
Fontaine, E. M., Devin, A., Rigoulet, M., & Leverve, X. M. (1997). The yield of oxidative phosphorylation is controlled both by force and flux. Biochemical and Biophysical Research Communications, 232(2), 532-5. [DOI:10.1006/bbrc.1997.6317] [PMID]
Garg, A., Bonanome, A., Grundy, S. M., Zhang, Z. J., & Unger, R. H. (1988). Comparison of a high-carbohydrate diet with a high-monounsaturated-fat diet in patients with non-insulin-dependent diabetes mellitus. The New England Journal of Medicine, 319(13), 829-34. [DOI:10.1056/NEJM198809293191304] [PMID]
Guo, Z., Chen, Y., Mao, Y. F., Zheng, T., Jiang, Y., & Yan, Y., et al. (2017). Long-term treatment with intranasal insulin ameliorates cognitive impairment, tau hyperphosphorylation, and microglial activation in a streptozotocin-induced Alzheimer’s rat model. Scientific Reports, 7, 45971. [DOI:10.1038/srep45971] [PMID] [PMCID]
Haas, R. H., Parikh, S., Falk, M. J., Saneto, R. P., Wolf, N. I., & Darin N., et al. (2007). Mitochondrial disease: A practical approach for primary care physicians. Pediatrics, 120(6), 1326-33. [DOI:10.1542/peds.2007-0391] [PMID]
Hafner, R. P., Brown, G. C., & Brand, M. D. (1990). Analysis of the control of respiration rate, phosphorylation rate, proton leak rate and protonmotive force in isolated mitochondria using the ‘top-down’ approach of metabolic control theory. European Journal of Biochemistry, 188(2), 313-9. [DOI:10.1111/j.1432-1033.1990.tb15405.x] [PMID]
Hindfelt, B., Plum, F., & Duffy, T. E. (1977). Effect of acute ammonia intoxication on cerebral metabolism in rats with portacaval shunts. The Journal of Clinical Investigation, 59(3), 386-96. [DOI:10.1172/JCI108651] [PMID] [PMCID]
Hirokawa, N., Noda, Y., Tanaka, Y., & Niwa, Sh. (2009). Kinesin superfamily motor proteins and intracellular transport. Nature Reviews Molecular Cell Biology Volume, 10(10), 682-96. [DOI:10.1038/nrm2774] [PMID]
Hollenbeck, P. J., & Saxton, W. M. (2005). The axonal transport of mitochondria. Journal of Cell Science, 118(Pt 23), 5411-9. [DOI:10.1242/jcs.02745] [PMID] [PMCID]
Hritcu, L., Cioanca, O., & Hancianu, M. (2012). Effects of lavender oil inhalation on improving scopolamine-induced spatial memory impairment in laboratory rats. Phytomedicine, 19(6), 529-34. [DOI:10.1016/j.phymed.2012.02.002] [PMID]
Huang, M., Camara, A. K., Stowe, D. F., Qi, F., & Beard, D. A. (2007). Mitochondrial inner membrane electrophysiology assessed by rhodamine-123 transport and fluorescence. Annals of Biomedical Engineering, 35(7), 1276-85. [DOI:10.1007/s10439-007-9265-2] [PMID] [PMCID]
Kamboj, S. S., Chopra, K., & Sandhir, R. (2008). Neuroprotective effect of N-acetylcysteine in the development of diabetic encephalopathy in streptozotocin-induced diabetes. Metabolic Brain Disease, 23(4), 427. [DOI:10.1007/s11011-008-9104-7] [PMID]
King, S. J., & Schroer, T. A. (2000). Dynactin increases the processivity of the cytoplasmic dynein motor. Nature Cell Biology, 2(1), 20-4. [DOI:10.1038/71338] [PMID]
Leszek, J., Trypka, E., Tarasov, V. V., Ashraf, G. M., & Aliev, G. (2017). Type 3 diabetes mellitus: A novel implication of Alzheimers disease. Current Topics in Medicinal Chemistry, 17(12), 1331-5. [DOI:10.2174/1568026617666170103163403] [PMID]
Letai, A. (2006). Growth factor withdrawal and apoptosis: The middle game. Molecular Cell, 21(6), 728-30. [DOI:10.1016/j.molcel.2006.03.005] [PMID]
Lu, T., Pan, Y., Kao, S. Y., Li, C., Kohane, I., & Chan, J., et al. (2004). Gene regulation and DNA damage in the ageing human brain. Nature, 429(6994), 883-91. [DOI:10.1038/nature02661] [PMID]
Lulat, S. I., Yadav, Y. C., Balaraman, R., & Maheshwari, R. (2016). Antiurolithiatic effect of lithocare against ethylene glycol-induced urolithiasis in Wistar rats. Indian Journal of Pharmacology, 48(1), 78-82. [DOI:10.4103/0253-7613.174564] [PMID] [PMCID]
Lupien, S. B., Bluhm, E. J., & Ishii, D. N. (2003). Systemic insulin-like growth factor-I administration prevents cognitive impairment in diabetic rats, and brain IGF regulates learning/memory in normal adult rats. Journal of Neuroscience Research, 74(4), 512-23. [DOI:10.1002/jnr.10791] [PMID]
Maassen, J. A., ‘t Hart, L. M., van Essen, E., Heine, R. J., Nijpels, G., & Jahangir Tafrechi, R. S., et al. (2004). Mitochondrial diabetes: Molecular mechanisms and clinical presentation. Diabetes, 53(Suppl 1), S103-9. [DOI:10.2337/diabetes.53.2007.S103] [PMID]
Maassen, J. A., Janssen, G. M. C., & ‘t Hart, L. M. (2005). Molecular mechanisms of mitochondrial diabetes (MIDD). Annals of Medicine, 37(3), 213-21. [DOI:10.1080/07853890510007188] [PMID]
McNay, E. C., & Recknagel, A. K. (2011). Brain insulin signaling: A key component of cognitive processes and a potential basis for cognitive impairment in type 2 diabetes. Neurobiology of Learning and Memory, 96(3), 432-42. [DOI:10.1016/j.nlm.2011.08.005] [PMID] [PMCID]
Mingatto, F. E., Rodrigues, T., Pigoso, A. A., Uyemura, S. A., Curti, C., & Santos, A. C. (2002). The critical role of mitochondrial energetic impairment in the toxicity of nimesulide to hepatocytes. Journal of Pharmacology and Experimental Therapeutics, 303(2), 601-7. [DOI:10.1124/jpet.102.038620] [PMID]
Miyata, Sh., Yamada, N., Hirano, S., Tanaka, S. I., & Kamei, J. (2007). Diabetes attenuates psychological stress-elicited 5-HT secretion in the prefrontal cortex but not in the amygdala of mice. Brain Research, 1147, 233-9. [DOI:10.1016/j.brainres.2007.02.001] [PMID]
Mousavi, F., Eidi, A., Khalili, M., & Roghani, M. (2018). Metformin ameliorates learning and memory deficits in streptozotocin-induced diabetic rats. Journal of Basic and Clinical Pathophysiology, 6(1), 17-22. [DOI:10.22070/JBCP.2017.2782.1085]
Musen, G., Lyoo, I. K., Sparks, C. R., Weinger, K., Hwang, J., & Ryan, C. M., et al. (2006). Effects of type 1 diabetes on gray matter density as measured by voxel-based morphometry. Diabetes, 55(2), 326-33. [DOI:10.2337/diabetes.55.02.06.db05-0520] [PMID]
Pellow, Sh., Chopin, P., File, S. E., & Briley, M. (1985). Validation of open: Closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. Journal of Neuroscience Methods, 14(3), 149-67. [DOI:10.1016/0165-0270(85)90031-7]
Pilling, A. D., Horiuchi, D., Lively, C. M., & Saxton, W. M. (2006). Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Molecular Biology of the Cell, 17(4), 2057-68. [DOI:10.1091/mbc.e05-06-0526] [PMID] [PMCID]
Pittenger, C., & Duman, R. S. (2008). Stress, depression, and neuroplasticity: A convergence of mechanisms. Neuropsychopharmacology, 33(1), 88-109. [DOI:10.1038/sj.npp.1301574] [PMID]
Prodanov, D., & Feirabend, H. K. P. (2007). Morphometric analysis of the fiber populations of the rat sciatic nerve, its spinal roots, and its major branches. Journal of Comparative Neurology, 503(1), 85-100. [DOI:10.1002/cne.21375] [PMID]
Rajashree, R., Kholkute, S. D., & Goudar, S. S. (2011). Effects of duration of diabetes on behavioural and cognitive parameters in streptozotocin-induced juvenile diabetic rats. The Malaysian Journal of Medical Sciences, 18(4), 26-31. [PMID] [PMCID]
Reske-Nielsen, E., Lundbaek, K., & Rafaelsen, O. J. (1966). Pathological changes in the central and peripheral nervous system of young long-term diabetics: I. diabetic encephalopathy. Diabetologia, 1(3-4), 233-41. [DOI:10.1007/BF01257917] [PMID]
Rivera, E. J., Goldin, A., Fulmer, N., Tavares, R., Wands, J. R., & de la Monte, S. M. (2005). Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. Journal of Alzheimer’s Disease, 8(3), 247-68. [DOI:10.3233/JAD-2005-8304] [PMID]
Roghani, M., Joghataie, M. T., Jalali, M. R., & Baluchnejadmojarad, T. (2006). Time course of changes in passive avoidance and Y-maze performance in male diabetic rats. Iranian Biomedical Journal, 10(2), 99-104. http://ibj.pasteur.ac.ir/article-1-355-en.html
Russo, G. J., Louie, K., Wellington, A., Macleod, G. T., Hu, F., & Panchumarthi, S., et al. (2009). Drosophila Miro is required for both anterograde and retrograde axonal mitochondrial transport. The Journal of Neuroscience, 29(17), 5443-55. [DOI:10.1523/JNEUROSCI.5417-08.2009] [PMID] [PMCID]
Sima, A. A. F., & Li, Z. G. (2005). The effect of C-peptide on cognitive dysfunction and hippocampal apoptosis in type 1 diabetic rats. Diabetes, 54(5), 1497-505. [DOI:10.2337/diabetes.54.5.1497] [PMID]
Sima, A. A. F., Zhang, W., Muzik, O., Kreipke, C. W., Rafols, J. A., & Hoffman, W. H. (2009). Sequential abnormalities in type 1 diabetic encephalopathy and the effects of C-Peptide. The Review of Diabetic Studies, 6(3), 211-22. [DOI:10.1900/RDS.2009.6.211] [PMID] [PMCID]
Steen, E., Terry, B. M., Rivera, E. J., Cannon, J. L., Neely, T. R., & Tavares, R., et al. (2005). Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease -- is this type 3 diabetes? Journal of Alzheimer’s Disease, 7(1), 63-80. [DOI:10.3233/JAD-2005-7107] [PMID]
Stewart, R., & Liolitsa, D. (1999). Type 2 diabetes mellitus, cognitive impairment and dementia. Diabetic Medicine, 16(2), 93-112. [DOI:10.1046/j.1464-5491.1999.00027.x] [PMID]
Strachan, M. W. J. (2011). R D Lawrence Lecture 2010. The brain as a target organ in type 2 diabetes: Exploring the links with cognitive impairment and dementia. Diabetic Medicine, 28(2), 141-7. [DOI:10.1111/j.1464-5491.2010.03199.x] [PMID]
Stranahan, A. M., Arumugam, T. V., Cutler, R. G., Lee, K., Egan, J. M., & Mattson, M. P. (2008). Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nature Neuroscience, 11(3), 309-17. [DOI:10.1038/nn2055] [PMID] [PMCID]
Tang, Z. J., Zou, W., Yuan, J., Zhang, P., Tian, Y., & Xiao, Z. F., et al. (2015). Antidepressant-like and anxiolytic-like effects of hydrogen sulfide in streptozotocin-induced diabetic rats through inhibition of hippocampal oxidative stress. Behavioural Pharmacology, 26(5), 427-35. [DOI:10.1097/FBP.0000000000000143] [PMID]
Wang, A., Lucero, E., Bennett, S., & Potter, H. (2016). O4‐03‐04: Type 3 diabetes in Alzheimer’s disease: Ab‐mediated inhibition of eg5/kinesin 5 leads to insulin receptor and glucose transporter mis‐localization and dysfunction. Alzheimer’s & Dementia, 12(7S Pt 7), 338. [DOI:10.1016/j.jalz.2016.06.621]
Zalsman, G., Huang, Y. Y., Oquendo, M. A., Burke, A. K., Hu, X. Z., & Brent, D. A., et al. (2006). Association of a triallelic serotonin transporter gene promoter region (5-HTTLPR) polymorphism with stressful life events and severity of depression. The American Journal of Psychiatry, 163(9), 1588-93. [DOI:10.1176/ajp.2006.163.9.1588] [PMID]
Zhao, C., Takita, J., Tanaka, Y., Setou, M., Nakagawa, T., & Takeda, S., et al. (2001). Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell, 105(5), 587-97. [DOI:10.1016/S0092-8674(01)00363-4]
Zilliox, L. A., Chadrasekaran, K., Kwan, J. Y., & Russell, J. W. (2016). Diabetes and cognitive impairment. Current Diabetes Reports, 16(9), 87. [DOI:10.1007/s11892-016-0775-x] [PMID] [PMCID]
Type of Study: Original |
Subject:
Cellular and molecular Neuroscience
Received: 2018/12/30 | Accepted: 2019/05/13 | Published: 2020/11/1
Received: 2018/12/30 | Accepted: 2019/05/13 | Published: 2020/11/1
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
