

Volume 10, Issue 6 (November & December 2019)
BCN 2019, 10(6): 545-556 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Amiri S, Azadmaneshb K, Dehghan Shasaltaneh M, Khoshkholgh-Sima B, Naghdi N. Protein Kinase Cε in the Platelet and Hippocampal Tissue as a Diagnostic Biological Marker in Alzheimer Disease. BCN 2019; 10 (6) :545-556
URL: http://bcn.iums.ac.ir/article-1-1376-en.html
URL: http://bcn.iums.ac.ir/article-1-1376-en.html
Sara Amiri1 

 , Kayhan Azadmaneshb2 , Marzieh Dehghan Shasaltaneh3 , Baharak Khoshkholgh-Sima1 , Nasser Naghdi *1
, Kayhan Azadmaneshb2 , Marzieh Dehghan Shasaltaneh3 , Baharak Khoshkholgh-Sima1 , Nasser Naghdi *1
, Kayhan Azadmaneshb2 , Marzieh Dehghan Shasaltaneh3 , Baharak Khoshkholgh-Sima1 , Nasser Naghdi *1
1- Department of Physiology and Pharmacology, Pasteur Institute of Iran, Tehran, Iran.
2- Department of Virology, Pasteur Institute of Iran, Tehran, Iran.
3- Department of Biology, Faculty of Sciences, University of Zanjan, Zanjan, Iran.
2- Department of Virology, Pasteur Institute of Iran, Tehran, Iran.
3- Department of Biology, Faculty of Sciences, University of Zanjan, Zanjan, Iran.
Full-Text [PDF 897 kb]
| Abstract (HTML)
Full-Text:
1. Introduction
Alzheimer disease (AD) is one of the most common neurodegenerative disorders. It is characterized by the loss of mental, behavioral, and functional abilities. Formation of amyloid plaques, tau hyperphosphorylation, and inflammation lead to synaptic impairment and destroying the integrity of brain functions (Kumar, Singh, & Ekavali, 2015). Pyramidal cells of the entorhinal cortex and the CA1 region of the hippocampus are destructed in Alzheimer brains (Huang & Mucke, 2012), which may be resulted from the deficiency of several enzymes in the mentioned regions, such as protein kinase C (PKC) and mitogen-activated protein kinase (MAPK) (Hong-Qi, Zhi-Kun, & Sheng-Di, 2012). PKC alters in fibroblasts, lymphocytes and red blood cells of AD patients; therefore, based on the previous reports, PKC conformation in peripheral cells can be an early predictive marker for AD (Humpel, 2011).
PKC isoenzymes increase during the associative learning and memory processes. Synaptogenic pathways are activated during the enhancement of PKC, which plays a critical role in associative learning and the regulation of synaptic and memory functions (Sun, Nelson, & Alkon, 2015).
An increase in the enzyme’s apparent affinity for Ca+2 and membrane phospholipids leads to stimulate PKC. Diacylglycerol (DAG) is generated by receptor-mediated hydrolysis of phosphoinositides in the PKC. The stimulation of PKC by its substrate like DAG translocates the enzyme from the cytosol to the specific location of neuronal tissues (Wang, Pisano, & Friedman, 1994).
The deficits in proper PKC translocation worsen stroke outcome and amyloid beta (Aβ) toxicity. PKC isoforms or “memory kinases” contribute to cognitive decline and its alteration by aging and AD progression (Sun & Alkon, 2014). They phosphorylate several proteins in the signaling pathway, like tau protein; therefore, it is identified as tau kinase. Tau hyperphosphorylation and phosphorylation of glycogen synthase kinase 3β (GSK-3β) are considered as one of the critical functions of PKC (De Montigny, Elhiri, Allyson, Cyr, & Massicotte, 2013).
PKC α, ε, and δ isoenzymes are associated with the α-secretase activity, increasing the non-toxic soluble amyloid precursor protein-α (s-APPα), and indirectly reducing the toxic Aβ as the degeneration product of β-secretase-mediate cleavage of APP (Khan & Alkon, 2010). Repression of the PKC gene has been linked to impaired memory and learning and their associated neurodegenerative diseases caused by several neurotransmitters, such as glutamate, dopamine, acetylcholine, and serotonin.
PKCε is one of the most critical isoenzymes involved in memory functions at several levels, such as post-translational modification of synaptic proteins, transcriptional activity, and local protein synthesis in synapses (Sun et al., 2015). The tertiary structure of PKCε includes a catalytic domain and two C1 domains with a direct translocation from the plasma membrane to the nuclear membrane. PKCε is associated with the regulation of memory and wound healing (Lucke-Wold et al., 2015). It is necessary for spatial memory formation and object recognition through functional reduction of Aβ accumulation and inducing the endothelin-converting enzyme to degrade Aβ40 and Aβ42 to small fragments (Pacheco-Quinto & Eckman, 2013).
The neuroprotective role of PKCε is through its translocation to synaptic membrane; therefore, it can be protective against memory decline in AD (Lucke-Wold et al., 2015). The loss of synaptogenic action of PKCε, its protective effects against neurogenic factors, lower levels of PKCε in the hippocampal area of AD brain are significant markers to distinguish AD from other disorders (Hongpaisan, Sun, & Alkon, 2011; Nelson, Sun, Hongpaisan, & Alkon, 2008). PKC and synaptic growth factors, like brain-derived neurotrophic factor (BDNF), are associated with synaptic regulation and replacement with a vital role at the core of the disease-modifying therapeutics (Sun et al., 2015). The high abundance of PKCε in presynaptic nerve fibers demonstrates a function in neurite outgrowth, synapse formation, and neurotransmitter release (Hongpaisan et al., 2011).
Phospholipid-dependent PKC activities and levels decrease in leukocytes and platelets of AD patients compared with the controls (Bosman et al., 1992; Lanius et al., 1997; Matsushima et al., 1994; Molchan et al., 1993). Low-density platelet populations reveal the increased serotonin content in dementia of the Alzheimer type (Milovanovic et al., 2014). Platelets provide APP as a precursor Aβ protein, so they are probably involved in AD pathophysiology. One of the features of progressive AD is malfunctioning platelets in dementia (Jaremo, Milovanovic, Buller, Nilsson, & Winblad, 2012). Human platelets contain high levels of APP and the concentrations of platelets are equivalent to the number of APP isoforms in the brain (Catricala, Torti, & Ricevuti, 2012).
Platelet agonists activate specific signaling pathways, such as different molecules and enzymes. Therefore, they can evoke a transient enhancement of intracellular Ca+2 concentrations (Catricala et al., 2012). The metabolism of platelet APP might also contribute to the accumulation of Aβ in the brain (Deane & Zlokovic, 2007; Roher et al., 2009). In the present study, we investigated the alteration of PKCε in the platelet and hippocampal tissue in the pathological diseases, like AD as a biological marker to detect the primary stages of the AD.
2. Methods
2.1. Animals
Adult male Wistar rats weighting 200-230 g were purchased from the Pasteur Institute of Iran and housed 4 per cage in large cages. Food and water were available, and the animals maintained at room temperature (24±1ºC) with a 12/12 h light/dark cycle (lights on at 7:00 AM). All efforts were made to reduce the number of testing on animals and their suffering during the experiments. After a week of adaptation to the room conditions, the rats were randomly divided into three groups (n=8) of the control (intact), sham (vehicle), and experimental. The experimental group was subjected to a bilateral injection of Aβ into the CA1 region, and because no significant difference was observed between the sham and control groups, in all experiments, the sham group was compared with the Aβ-injected group.
2.2. Drug administration
Human Aβ (1-42) (Tocris, UK) was dissolved in sterile deionized water (vehicle) at a concentration of 5 μg/μL and incubated at 37o C for 4 days to obtain the aggregated form. The animals were anesthetized with a combination of ketamine (50 mg/kg, IP) and xylazine (5 mg/kg, IP) and placed in a stereotaxic apparatus (Stoelting, Wood Dale, IL, USA). One microliter Aβ or vehicle (sham group) was infused bilaterally into the CA1 region of the hippocampus (1 µL/CA1) according to the rat brain atlas of Paxinos and Watson (Paxinos & Watson, 1986) with the following coordinates: AP -3.80 mm from bregma, ML ±2.2 mm from midline, and DV -2.7 mm from the skull surface. The rate of injection was 0.5 μL/min. After completion of the infusion, the needle was left in place for 3 min to allow diffusion of the drug from the needle.
2.3. Behavioral assessment
The Morris water maze (MWM) test was performed in a water tank according to a previous procedure conducted in our laboratory. In short, animals received a block of 4 trials during 5 daily sessions. During the first 4 days (on days 6, 7, 8 and 9 after Aβ injection), the platform site did not change throughout training and situated in the center of the southwest quadrant, but animals were released randomly into the water (while facing to the tank wall) from a different location (north, east, south, and west) between the trials. The platform was submerged 1.5 cm below the surface of the water for testing spatial learning and memory. A trial was started by placing a rat into the pool. Each of four starting positions was used once in a series of four trials. Each trial was terminated as soon as the rat had climbed onto the escape platform or when 90 s had elapsed. A rat was allowed to stay on the platform for 20 s. Then, it was taken from the platform, and the next trial was started. If rats were not able to find the platform after 90 s, they would be put on the platform by the experimenter and allowed to rest on it for 20 s. At the end of the fourth trial, rats were gently dried with a towel, kept warm for an hour and returned to their home cages.
The path of each rat in each trial was automatically recorded by a computerized system and then analyzed by several parameters, such as escape latency to find the platform and traveled distance. All experiments were conducted between 10:00 AM and 1:00 PM. For each animal, two factors were evaluated: the time spent to find the platform (escape latency, s) and the distance traveled before finding the platform (cm).
Another assessment was done by visual and spatial probe test. On day 5 of behavioral assessment (ten days after Aβ injection), the platform was elevated above the water surface, covered by bright color aluminum foil, and placed in the center of the southeast quadrant, which assessed the motivation and sensorimotor coordination toward a visible platform. The spatial probe test was also performed on day 5, in which the hidden platform was removed from the target quadrant and rats were allowed to swim for 90 s. The rats were released in the water in a location that was exactly opposite from where the platform was placed. Their behavior was recorded with a video tracking system and also escape latencies were noted for further analysis.
2.4. Sample collection and processing
Blood samples were collected on days 0, 5, 10, and 30 after injection of Aβ or solvent and also from the control (intact) group and then the animals were decapitated. The brains were removed, and then hippocampus was separated.
2.5. Histological evaluation
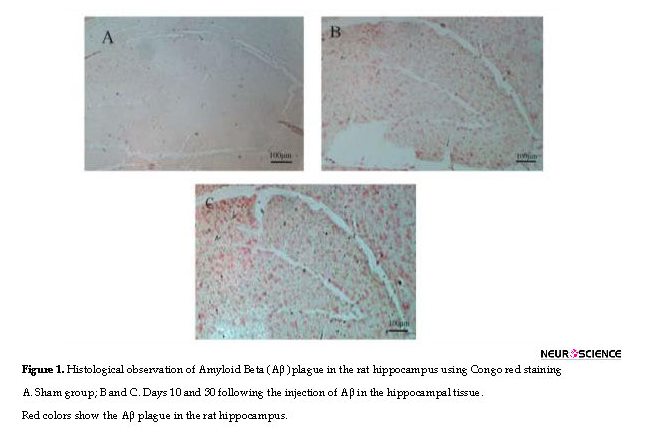
One rat from each Aβ-injected group (10 and 30 days) was anesthetized and then transcardially perfused with phosphate-buffered saline followed by 4% (w/v) formaldehyde. Brains were rapidly removed and fixed in 30% (v/v) buffered formalin, embedded with paraffin, cut into consecutive 5-μm transverse sections with a microtome and placed on poly-d-lysine-coated glass slides. The sections were stained with Congo red to observe Aβ plaques.
2.6. Blood sample and platelet isolation
Rats were anesthetized with ether and blood samples were collected into a tube containing anticoagulant acid citrate dextrose. They were centrifuged immediately at 2300×g to obtain Platelet-Rich Plasma (PRP). The duration of centrifugation is associated with the volume (10 s/mL). The PRP was centrifuged at 2200×g for 8 min to obtain the platelet pellet. The plasma was removed and the platelets were resuspended in Tyrode’s buffer (10 mM HEPES, 0.4 mM NaH2PO4, 137 mM NaCl, 5.5 mM glucose, 2.8 mM KCl, 1 mM MgCl, and 12 mM NaHCO3) containing heparin (10 U/mL) and PGI2 (0.5 μM). After incubation for 10 min at 37°C, PGI2 (0.5 μM) was added and depending on the suspension volume, centrifuged at 1900×g. The buffer was removed and the platelets resuspended again in Tyrode’s buffer containing 0.5 μM PGI2 and after incubation for 10 min at 37°C, it was centrifuged for the second time. The platelet pellet was finally frozen and stored at -80°C until use.
2.7. The PKC activity assay
Tissue samples and platelets for PKC activity assay were prepared. The hippocampus and platelet pellet were homogenized in lysis buffer containing Tris-HCl (20 mM; pH 7.4), EGTA (2 mM), EDTA (5 mM), 0.2% Triton X-100, dithiothreitol (1 mM), protease inhibitor cocktail (P8340), and phosphatase inhibitor cocktail (P5726) (Sigma-Aldrich). Homogenates were centrifuged at 14,000 g for 10 min to remove debris. The concentration of protein was determined using the QuantiPro BCA assay kit (Sigma-Aldrich).
PKC activity was measured using a kit according to the manufacturer’s instructions (ADI-EKS-420A, Enzo Life Sciences), which is based on a solid-phase enzyme-linked immunosorbent assay. It utilizes a specific synthetic peptide as a substrate for subtypes of PKCs and a polyclonal antibody for the phosphorylated form of the substrate. The absorbance was determined at a wavelength of 450 nm, and PKC activity was expressed as a relative activity. Data were normalized to total protein content (μg), as measured by the BCA assay.
2.8. RNA extraction
Total RNA was extracted from the hippocampus and isolated platelets using the TRIzol reagent (Invitrogen), based on the manufacturer’s instructions. Briefly, after homogenizing by TRIzol reagent, chloroform (Merck, Germany) was added to the solution and centrifuged for 15 min at 12000×g at 4°C. The upper phase was then transferred into another tube, and RNA was precipitated with isopropanol (Merck) at 4°C. The mixture was centrifuged for 15 min at 12000×g at 4°C. The resulting pellet was then washed in 75% (v/v) ethanol and dissolved in diethylpyrocarbonate (DEPC)-treated water. The concentration of the isolated RNA was determined with the Picodrop spectrophotometer.
2.9. Complementary DNA (cDNA) synthesis
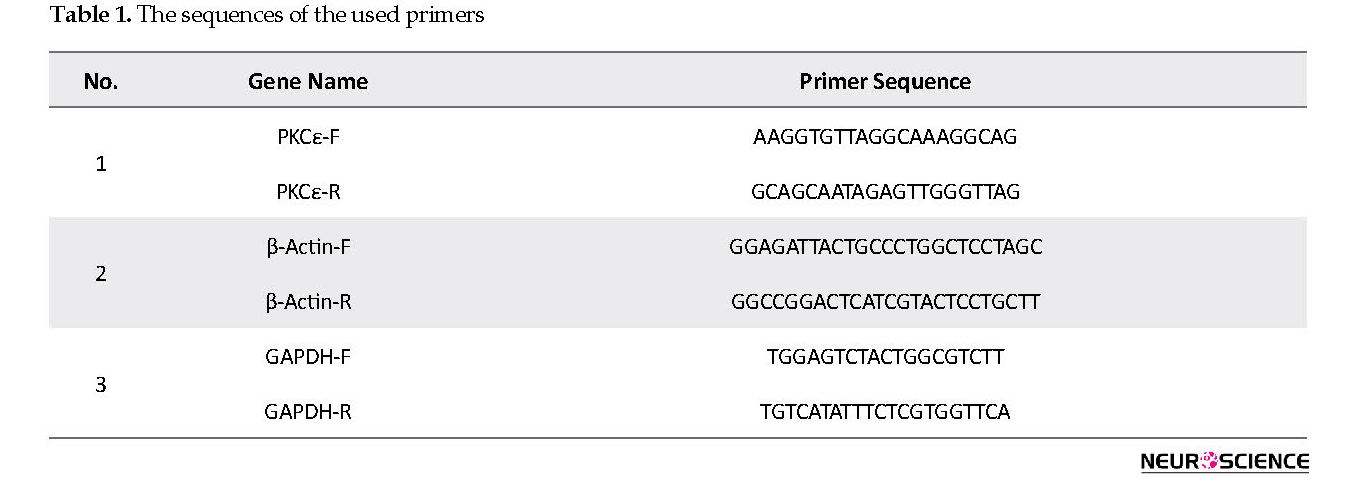
Complementary DNA (cDNA) synthesis was carried out using 1 μg of RNA and RevertAid RT (200 U/μL) with oligo(dT)18 (Thermo scientific) priming in a 20 μL reaction according to the manufacturer’s instructions. Rat specific primers for PKCε, β-actin, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were synthesized as HPLC grade. All used primers were blasted against the rat genome to ensure that they are not complementary to other regions of the genome. Table 1 presents the used primers.
2.10. Quantitative real-time PCR (RT-qPCR)
PKCε expression was assessed by RT-qPCR using SYBR Premix Ex Taq II (TAKARA). For each subject, the PCR reaction was performed in triplicate in a final volume of 20 μL using 2 μL of synthesized cDNA. The PCR reactions were performed on ABI 7500 fast (Applied Biosystems) with the recommended cycling temperatures provided in the TAKARA guide.
The relative expression software tool 2009 (REST, QIAGEN) was used to compare the PKCε expression of the Aβ-treated and sham groups in the hippocampus and platelet samples. The expression ratio of PKCε was measured and the results were normalized by the GAPDH and β-actin mRNA as the reference genes. Serial dilutions were done to create a standard curve and threshold cycle numbers were averaged.
2.11. Quantitation of PKCε by Western Blotting
The hippocampus and isolated platelets were homogenized in the lysis buffer containing 1 mmol/L EDTA, 150 mmol/L NaCl, 1% (v/v) Triton X-100, 50 mmol/L Tris-HCl, 0.1% (v/v) SDS (pH 8), and protease inhibitors cocktails. Total proteins were electrophoresed in 12% SDS-PAGE gels, transferred to Polyvinylidene fluoride membranes (PVDF), incubated with anti-PKCε (C-15; Santa Cruz, 1/500 [v/v] and 1/1000 [v/v] dilution for platelet and hippocampus, respectively) and anti-β-actin (R-22; Santa Cruz, 1/1000 [v/v] dilution) antibodies followed by enhanced chemiluminescence detection (ECL; Amersham). Data analysis was done using Image J after background subtraction, and the densities of PKCε bands were measured and their ratios to β-actin were evaluated. Protein concentration was determined using the QuantiPro BCA assay kit (Sigma-Aldrich).
Alzheimer disease (AD) is one of the most common neurodegenerative disorders. It is characterized by the loss of mental, behavioral, and functional abilities. Formation of amyloid plaques, tau hyperphosphorylation, and inflammation lead to synaptic impairment and destroying the integrity of brain functions (Kumar, Singh, & Ekavali, 2015). Pyramidal cells of the entorhinal cortex and the CA1 region of the hippocampus are destructed in Alzheimer brains (Huang & Mucke, 2012), which may be resulted from the deficiency of several enzymes in the mentioned regions, such as protein kinase C (PKC) and mitogen-activated protein kinase (MAPK) (Hong-Qi, Zhi-Kun, & Sheng-Di, 2012). PKC alters in fibroblasts, lymphocytes and red blood cells of AD patients; therefore, based on the previous reports, PKC conformation in peripheral cells can be an early predictive marker for AD (Humpel, 2011).
PKC isoenzymes increase during the associative learning and memory processes. Synaptogenic pathways are activated during the enhancement of PKC, which plays a critical role in associative learning and the regulation of synaptic and memory functions (Sun, Nelson, & Alkon, 2015).
An increase in the enzyme’s apparent affinity for Ca+2 and membrane phospholipids leads to stimulate PKC. Diacylglycerol (DAG) is generated by receptor-mediated hydrolysis of phosphoinositides in the PKC. The stimulation of PKC by its substrate like DAG translocates the enzyme from the cytosol to the specific location of neuronal tissues (Wang, Pisano, & Friedman, 1994).
The deficits in proper PKC translocation worsen stroke outcome and amyloid beta (Aβ) toxicity. PKC isoforms or “memory kinases” contribute to cognitive decline and its alteration by aging and AD progression (Sun & Alkon, 2014). They phosphorylate several proteins in the signaling pathway, like tau protein; therefore, it is identified as tau kinase. Tau hyperphosphorylation and phosphorylation of glycogen synthase kinase 3β (GSK-3β) are considered as one of the critical functions of PKC (De Montigny, Elhiri, Allyson, Cyr, & Massicotte, 2013).
PKC α, ε, and δ isoenzymes are associated with the α-secretase activity, increasing the non-toxic soluble amyloid precursor protein-α (s-APPα), and indirectly reducing the toxic Aβ as the degeneration product of β-secretase-mediate cleavage of APP (Khan & Alkon, 2010). Repression of the PKC gene has been linked to impaired memory and learning and their associated neurodegenerative diseases caused by several neurotransmitters, such as glutamate, dopamine, acetylcholine, and serotonin.
PKCε is one of the most critical isoenzymes involved in memory functions at several levels, such as post-translational modification of synaptic proteins, transcriptional activity, and local protein synthesis in synapses (Sun et al., 2015). The tertiary structure of PKCε includes a catalytic domain and two C1 domains with a direct translocation from the plasma membrane to the nuclear membrane. PKCε is associated with the regulation of memory and wound healing (Lucke-Wold et al., 2015). It is necessary for spatial memory formation and object recognition through functional reduction of Aβ accumulation and inducing the endothelin-converting enzyme to degrade Aβ40 and Aβ42 to small fragments (Pacheco-Quinto & Eckman, 2013).
The neuroprotective role of PKCε is through its translocation to synaptic membrane; therefore, it can be protective against memory decline in AD (Lucke-Wold et al., 2015). The loss of synaptogenic action of PKCε, its protective effects against neurogenic factors, lower levels of PKCε in the hippocampal area of AD brain are significant markers to distinguish AD from other disorders (Hongpaisan, Sun, & Alkon, 2011; Nelson, Sun, Hongpaisan, & Alkon, 2008). PKC and synaptic growth factors, like brain-derived neurotrophic factor (BDNF), are associated with synaptic regulation and replacement with a vital role at the core of the disease-modifying therapeutics (Sun et al., 2015). The high abundance of PKCε in presynaptic nerve fibers demonstrates a function in neurite outgrowth, synapse formation, and neurotransmitter release (Hongpaisan et al., 2011).
Phospholipid-dependent PKC activities and levels decrease in leukocytes and platelets of AD patients compared with the controls (Bosman et al., 1992; Lanius et al., 1997; Matsushima et al., 1994; Molchan et al., 1993). Low-density platelet populations reveal the increased serotonin content in dementia of the Alzheimer type (Milovanovic et al., 2014). Platelets provide APP as a precursor Aβ protein, so they are probably involved in AD pathophysiology. One of the features of progressive AD is malfunctioning platelets in dementia (Jaremo, Milovanovic, Buller, Nilsson, & Winblad, 2012). Human platelets contain high levels of APP and the concentrations of platelets are equivalent to the number of APP isoforms in the brain (Catricala, Torti, & Ricevuti, 2012).
Platelet agonists activate specific signaling pathways, such as different molecules and enzymes. Therefore, they can evoke a transient enhancement of intracellular Ca+2 concentrations (Catricala et al., 2012). The metabolism of platelet APP might also contribute to the accumulation of Aβ in the brain (Deane & Zlokovic, 2007; Roher et al., 2009). In the present study, we investigated the alteration of PKCε in the platelet and hippocampal tissue in the pathological diseases, like AD as a biological marker to detect the primary stages of the AD.
2. Methods
2.1. Animals
Adult male Wistar rats weighting 200-230 g were purchased from the Pasteur Institute of Iran and housed 4 per cage in large cages. Food and water were available, and the animals maintained at room temperature (24±1ºC) with a 12/12 h light/dark cycle (lights on at 7:00 AM). All efforts were made to reduce the number of testing on animals and their suffering during the experiments. After a week of adaptation to the room conditions, the rats were randomly divided into three groups (n=8) of the control (intact), sham (vehicle), and experimental. The experimental group was subjected to a bilateral injection of Aβ into the CA1 region, and because no significant difference was observed between the sham and control groups, in all experiments, the sham group was compared with the Aβ-injected group.
2.2. Drug administration
Human Aβ (1-42) (Tocris, UK) was dissolved in sterile deionized water (vehicle) at a concentration of 5 μg/μL and incubated at 37o C for 4 days to obtain the aggregated form. The animals were anesthetized with a combination of ketamine (50 mg/kg, IP) and xylazine (5 mg/kg, IP) and placed in a stereotaxic apparatus (Stoelting, Wood Dale, IL, USA). One microliter Aβ or vehicle (sham group) was infused bilaterally into the CA1 region of the hippocampus (1 µL/CA1) according to the rat brain atlas of Paxinos and Watson (Paxinos & Watson, 1986) with the following coordinates: AP -3.80 mm from bregma, ML ±2.2 mm from midline, and DV -2.7 mm from the skull surface. The rate of injection was 0.5 μL/min. After completion of the infusion, the needle was left in place for 3 min to allow diffusion of the drug from the needle.
2.3. Behavioral assessment
The Morris water maze (MWM) test was performed in a water tank according to a previous procedure conducted in our laboratory. In short, animals received a block of 4 trials during 5 daily sessions. During the first 4 days (on days 6, 7, 8 and 9 after Aβ injection), the platform site did not change throughout training and situated in the center of the southwest quadrant, but animals were released randomly into the water (while facing to the tank wall) from a different location (north, east, south, and west) between the trials. The platform was submerged 1.5 cm below the surface of the water for testing spatial learning and memory. A trial was started by placing a rat into the pool. Each of four starting positions was used once in a series of four trials. Each trial was terminated as soon as the rat had climbed onto the escape platform or when 90 s had elapsed. A rat was allowed to stay on the platform for 20 s. Then, it was taken from the platform, and the next trial was started. If rats were not able to find the platform after 90 s, they would be put on the platform by the experimenter and allowed to rest on it for 20 s. At the end of the fourth trial, rats were gently dried with a towel, kept warm for an hour and returned to their home cages.
The path of each rat in each trial was automatically recorded by a computerized system and then analyzed by several parameters, such as escape latency to find the platform and traveled distance. All experiments were conducted between 10:00 AM and 1:00 PM. For each animal, two factors were evaluated: the time spent to find the platform (escape latency, s) and the distance traveled before finding the platform (cm).
Another assessment was done by visual and spatial probe test. On day 5 of behavioral assessment (ten days after Aβ injection), the platform was elevated above the water surface, covered by bright color aluminum foil, and placed in the center of the southeast quadrant, which assessed the motivation and sensorimotor coordination toward a visible platform. The spatial probe test was also performed on day 5, in which the hidden platform was removed from the target quadrant and rats were allowed to swim for 90 s. The rats were released in the water in a location that was exactly opposite from where the platform was placed. Their behavior was recorded with a video tracking system and also escape latencies were noted for further analysis.
2.4. Sample collection and processing
Blood samples were collected on days 0, 5, 10, and 30 after injection of Aβ or solvent and also from the control (intact) group and then the animals were decapitated. The brains were removed, and then hippocampus was separated.
2.5. Histological evaluation
One rat from each Aβ-injected group (10 and 30 days) was anesthetized and then transcardially perfused with phosphate-buffered saline followed by 4% (w/v) formaldehyde. Brains were rapidly removed and fixed in 30% (v/v) buffered formalin, embedded with paraffin, cut into consecutive 5-μm transverse sections with a microtome and placed on poly-d-lysine-coated glass slides. The sections were stained with Congo red to observe Aβ plaques.
2.6. Blood sample and platelet isolation
Rats were anesthetized with ether and blood samples were collected into a tube containing anticoagulant acid citrate dextrose. They were centrifuged immediately at 2300×g to obtain Platelet-Rich Plasma (PRP). The duration of centrifugation is associated with the volume (10 s/mL). The PRP was centrifuged at 2200×g for 8 min to obtain the platelet pellet. The plasma was removed and the platelets were resuspended in Tyrode’s buffer (10 mM HEPES, 0.4 mM NaH2PO4, 137 mM NaCl, 5.5 mM glucose, 2.8 mM KCl, 1 mM MgCl, and 12 mM NaHCO3) containing heparin (10 U/mL) and PGI2 (0.5 μM). After incubation for 10 min at 37°C, PGI2 (0.5 μM) was added and depending on the suspension volume, centrifuged at 1900×g. The buffer was removed and the platelets resuspended again in Tyrode’s buffer containing 0.5 μM PGI2 and after incubation for 10 min at 37°C, it was centrifuged for the second time. The platelet pellet was finally frozen and stored at -80°C until use.
2.7. The PKC activity assay
Tissue samples and platelets for PKC activity assay were prepared. The hippocampus and platelet pellet were homogenized in lysis buffer containing Tris-HCl (20 mM; pH 7.4), EGTA (2 mM), EDTA (5 mM), 0.2% Triton X-100, dithiothreitol (1 mM), protease inhibitor cocktail (P8340), and phosphatase inhibitor cocktail (P5726) (Sigma-Aldrich). Homogenates were centrifuged at 14,000 g for 10 min to remove debris. The concentration of protein was determined using the QuantiPro BCA assay kit (Sigma-Aldrich).
PKC activity was measured using a kit according to the manufacturer’s instructions (ADI-EKS-420A, Enzo Life Sciences), which is based on a solid-phase enzyme-linked immunosorbent assay. It utilizes a specific synthetic peptide as a substrate for subtypes of PKCs and a polyclonal antibody for the phosphorylated form of the substrate. The absorbance was determined at a wavelength of 450 nm, and PKC activity was expressed as a relative activity. Data were normalized to total protein content (μg), as measured by the BCA assay.
2.8. RNA extraction
Total RNA was extracted from the hippocampus and isolated platelets using the TRIzol reagent (Invitrogen), based on the manufacturer’s instructions. Briefly, after homogenizing by TRIzol reagent, chloroform (Merck, Germany) was added to the solution and centrifuged for 15 min at 12000×g at 4°C. The upper phase was then transferred into another tube, and RNA was precipitated with isopropanol (Merck) at 4°C. The mixture was centrifuged for 15 min at 12000×g at 4°C. The resulting pellet was then washed in 75% (v/v) ethanol and dissolved in diethylpyrocarbonate (DEPC)-treated water. The concentration of the isolated RNA was determined with the Picodrop spectrophotometer.
2.9. Complementary DNA (cDNA) synthesis
Complementary DNA (cDNA) synthesis was carried out using 1 μg of RNA and RevertAid RT (200 U/μL) with oligo(dT)18 (Thermo scientific) priming in a 20 μL reaction according to the manufacturer’s instructions. Rat specific primers for PKCε, β-actin, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were synthesized as HPLC grade. All used primers were blasted against the rat genome to ensure that they are not complementary to other regions of the genome. Table 1 presents the used primers.
2.10. Quantitative real-time PCR (RT-qPCR)
PKCε expression was assessed by RT-qPCR using SYBR Premix Ex Taq II (TAKARA). For each subject, the PCR reaction was performed in triplicate in a final volume of 20 μL using 2 μL of synthesized cDNA. The PCR reactions were performed on ABI 7500 fast (Applied Biosystems) with the recommended cycling temperatures provided in the TAKARA guide.
The relative expression software tool 2009 (REST, QIAGEN) was used to compare the PKCε expression of the Aβ-treated and sham groups in the hippocampus and platelet samples. The expression ratio of PKCε was measured and the results were normalized by the GAPDH and β-actin mRNA as the reference genes. Serial dilutions were done to create a standard curve and threshold cycle numbers were averaged.
2.11. Quantitation of PKCε by Western Blotting
The hippocampus and isolated platelets were homogenized in the lysis buffer containing 1 mmol/L EDTA, 150 mmol/L NaCl, 1% (v/v) Triton X-100, 50 mmol/L Tris-HCl, 0.1% (v/v) SDS (pH 8), and protease inhibitors cocktails. Total proteins were electrophoresed in 12% SDS-PAGE gels, transferred to Polyvinylidene fluoride membranes (PVDF), incubated with anti-PKCε (C-15; Santa Cruz, 1/500 [v/v] and 1/1000 [v/v] dilution for platelet and hippocampus, respectively) and anti-β-actin (R-22; Santa Cruz, 1/1000 [v/v] dilution) antibodies followed by enhanced chemiluminescence detection (ECL; Amersham). Data analysis was done using Image J after background subtraction, and the densities of PKCε bands were measured and their ratios to β-actin were evaluated. Protein concentration was determined using the QuantiPro BCA assay kit (Sigma-Aldrich).
2.12. Statistical analysis
The obtained data were expressed as Mean±SEM and analyzed by one-way analysis of variance (ANOVA), followed by Tukey’s posthoc test. In all comparisons, P<0.05 was used as the criterion for statistical significance. In this study, t-test was used for comparison between the sham and treated groups following the injection of Aβ on days 0, 5, 10, and 30, separately.
3. Results
3.1. Histological study
The histological observation showed that Aβ plaques appeared in the rat brain slices after Aβ injection on days 10 and 30 (Figure 1).
3.2. The effect of Aβ injection on spatial memory of rats tested by the Morris water maze test
3.2.1. Acquisition tests
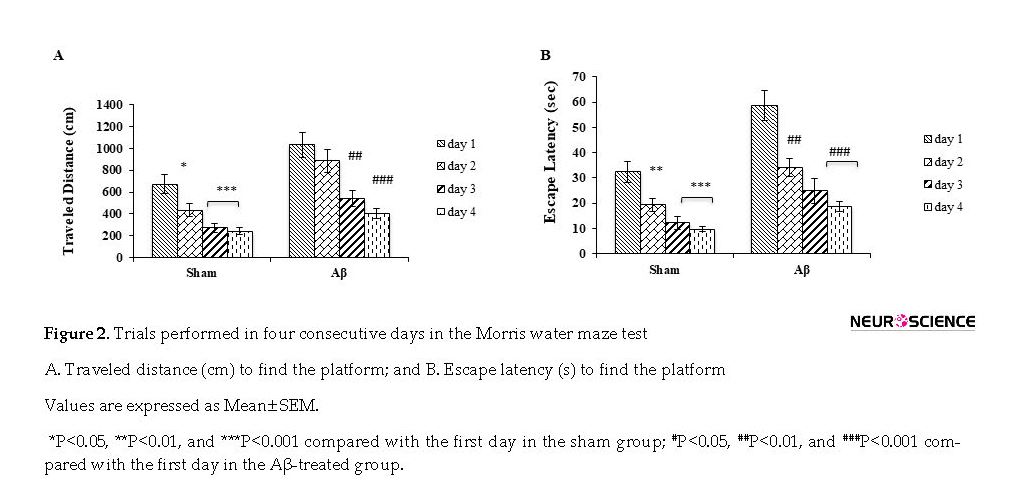
The traveled distance for finding the platform below the water surface during 4 days of acquisition trials was significantly longer in the Aβ-treated group (5 µg/µL) (F3,28=10.224; P<0.001) (Figure 2 A). Escape latency significantly decreased in Aβ-treated group (F3,28=13.476; P<0.001) for 4 days (Figure 2 B). There was a significant difference in terms of average escape latency (F3,28=19.508) and traveled distance (F3,28=13.441) in the treated group compared with the sham groups during 4 days of trials (P<0.001).
The obtained data were expressed as Mean±SEM and analyzed by one-way analysis of variance (ANOVA), followed by Tukey’s posthoc test. In all comparisons, P<0.05 was used as the criterion for statistical significance. In this study, t-test was used for comparison between the sham and treated groups following the injection of Aβ on days 0, 5, 10, and 30, separately.
3. Results
3.1. Histological study
The histological observation showed that Aβ plaques appeared in the rat brain slices after Aβ injection on days 10 and 30 (Figure 1).
3.2. The effect of Aβ injection on spatial memory of rats tested by the Morris water maze test
3.2.1. Acquisition tests
The traveled distance for finding the platform below the water surface during 4 days of acquisition trials was significantly longer in the Aβ-treated group (5 µg/µL) (F3,28=10.224; P<0.001) (Figure 2 A). Escape latency significantly decreased in Aβ-treated group (F3,28=13.476; P<0.001) for 4 days (Figure 2 B). There was a significant difference in terms of average escape latency (F3,28=19.508) and traveled distance (F3,28=13.441) in the treated group compared with the sham groups during 4 days of trials (P<0.001).
3.2.2. Probe trial test
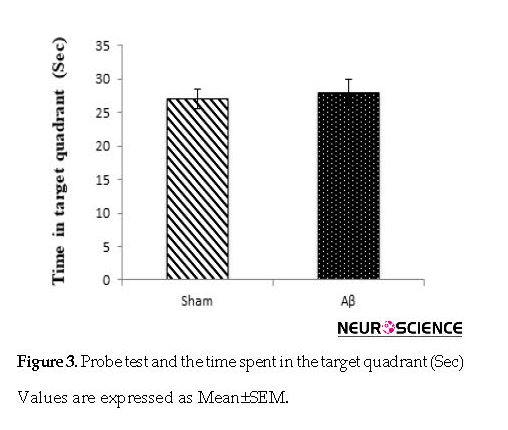
The results of the probe trial were evaluated based on the time spent in the target quadrant. No statistically significant differences were found in probe trial compared with the sham (P>0.05) and control (P>0.05, data are not shown) groups in escape latency onto the visible platform on day 5 (Figure 3).
The results of the probe trial were evaluated based on the time spent in the target quadrant. No statistically significant differences were found in probe trial compared with the sham (P>0.05) and control (P>0.05, data are not shown) groups in escape latency onto the visible platform on day 5 (Figure 3).
3.3. PKC activity
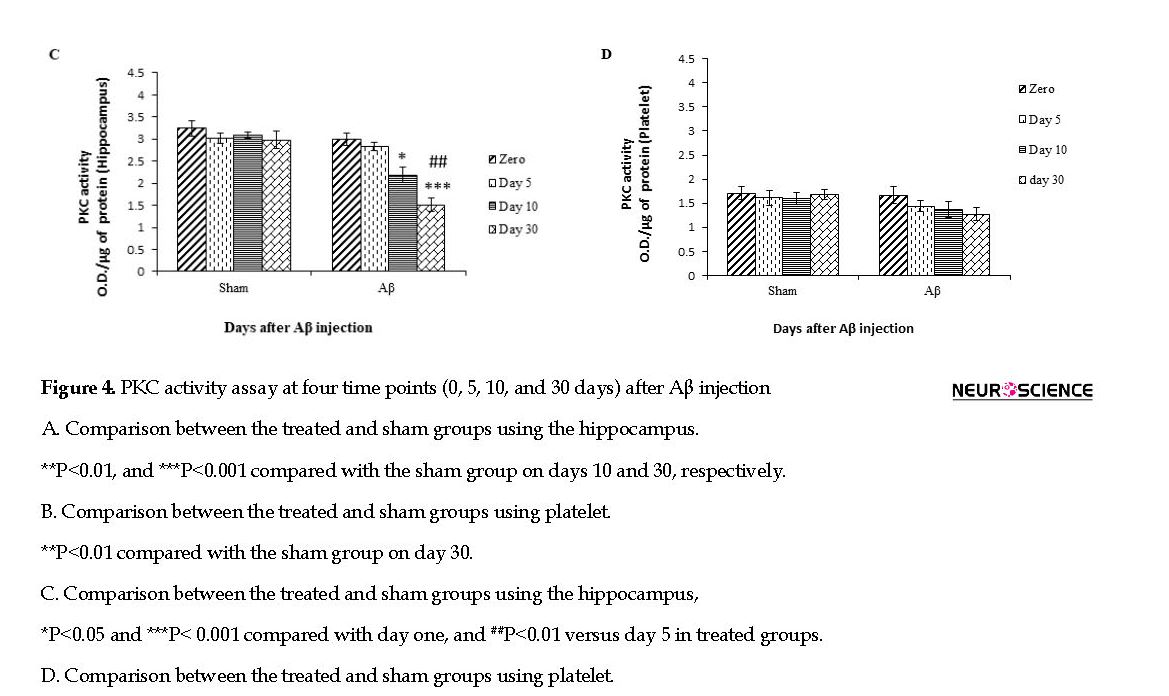
A significant difference was observed between the Aβ-treated and sham groups in terms of PKC activity in the hippocampus samples on days 10 and (P<0.01 and P<0.001, respectively) 30 following Aβ injection, but there was a significant difference in the PKC activity in the platelet sample between the Aβ-treated and sham groups on day 30 (P<0.01) after Aβ injection (Figure 4 A & B).
A significant difference was observed between the Aβ-treated and sham groups in terms of PKC activity in the hippocampus samples on days 10 and (P<0.01 and P<0.001, respectively) 30 following Aβ injection, but there was a significant difference in the PKC activity in the platelet sample between the Aβ-treated and sham groups on day 30 (P<0.01) after Aβ injection (Figure 4 A & B).
Also, there was a significant difference on the days 0 and 10 (F3,28=19.08; P<0.05), days 0 and 30 (F3,28=19.08; P<0.001), and days 5 and 30 (F3,28=19.08; P<0.01) following injection of Aβ in the hippocampal tissue of the treated groups, but no significant difference was observed in platelet samples using one-way ANOVA (Figure 4 C & D).
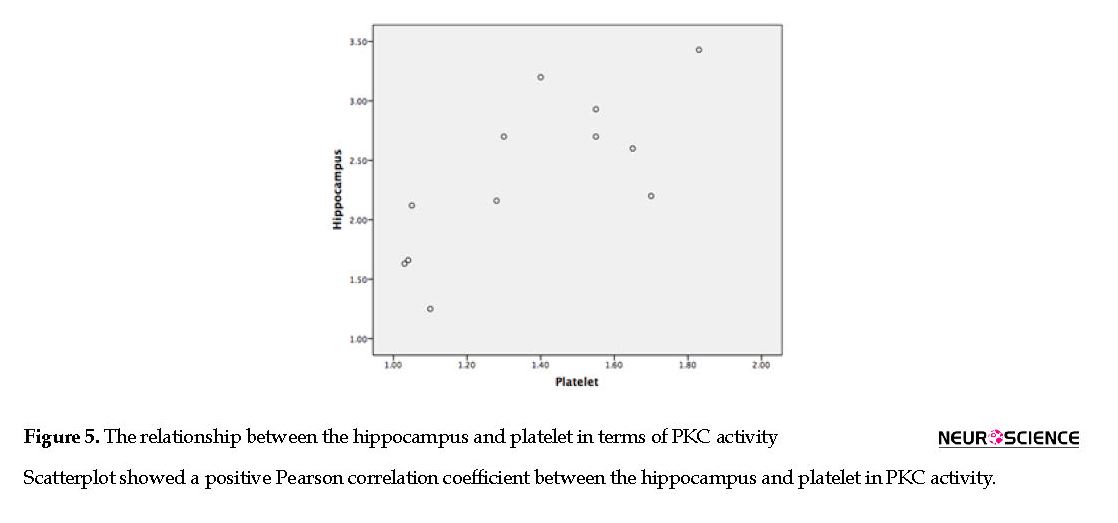
The Pearson product-moment correlation coefficient was used to assess the relationship between the hippocampus and platelet in PKC activity of the Aβ-treated group. Our results showed a positive correlation between the hippocampus and platelet in PKC activity (P<0.75; r=0.01) (Figure 5).
3.4. PKCε signaling in the hippocampus and platelet of the treated groups using real-time qPCR
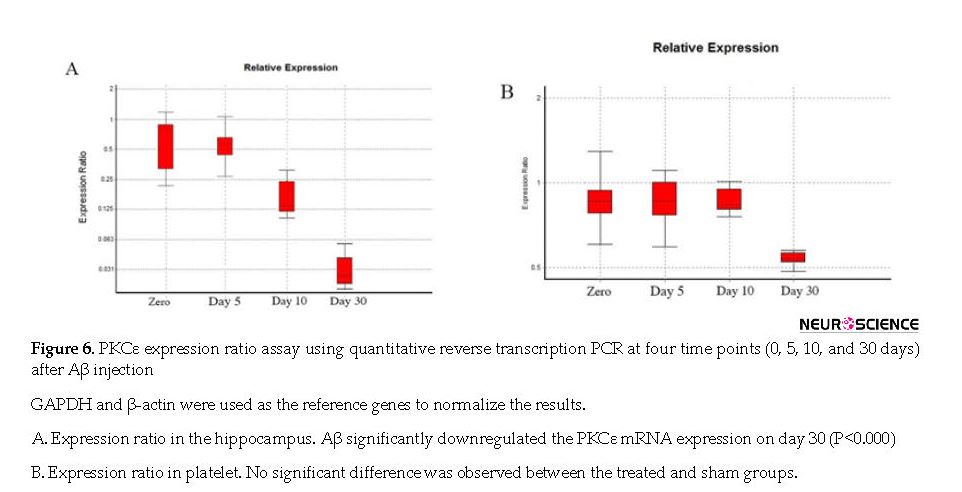
We studied the profile of PKCε expression by RT-qPCR in the hippocampus and platelet of Aβ-treated rats. The target sequence was confirmed by sequencing of PCR products. Aβ (5 µg/µL) significantly downregulated the PKCε mRNA expression in the hippocampus of rats on day 30 (P<0.000), but there was no significant difference in PKCε expression in platelet (P<0.059) (Figure 6 A & B).
We studied the profile of PKCε expression by RT-qPCR in the hippocampus and platelet of Aβ-treated rats. The target sequence was confirmed by sequencing of PCR products. Aβ (5 µg/µL) significantly downregulated the PKCε mRNA expression in the hippocampus of rats on day 30 (P<0.000), but there was no significant difference in PKCε expression in platelet (P<0.059) (Figure 6 A & B).
3.5. PKCε signaling in the hippocampus and platelet of the treated groups using western blotting analysis
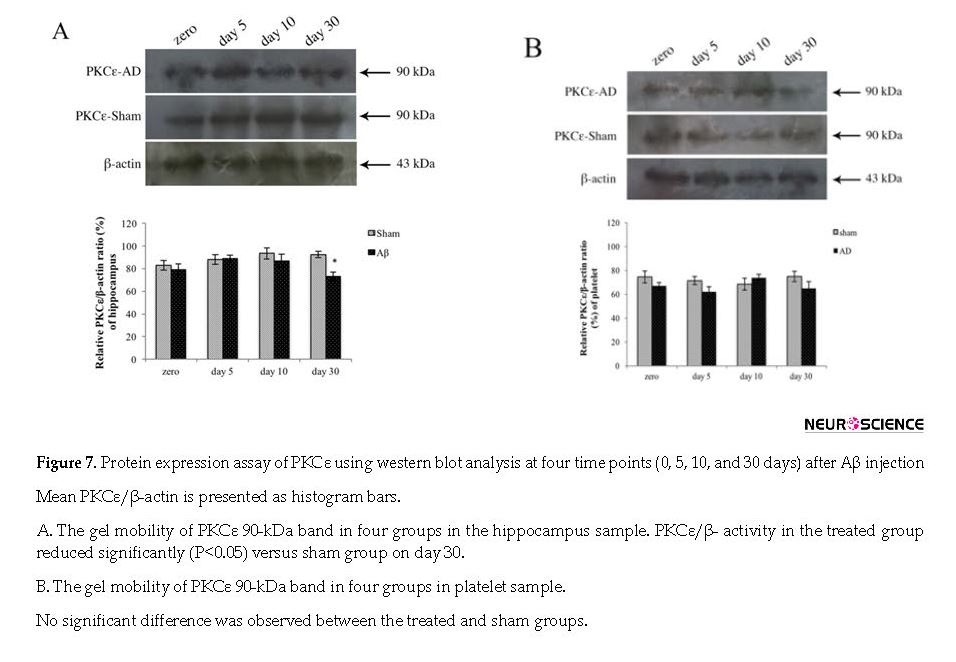
The effect of Aβ on PKCε protein expression was assessed with western blot analysis. There was no significant difference on the days 0, 5, 10, and 30 after the injection in the sham and also Aβ-treated groups using one-way ANOVA. Aβ significantly downregulated the PKCε protein expression in the hippocampus of treated groups on day 30 (P<0.05); however, there was no significant difference in PKCε expression in platelet sample (Figure 7 A & B).
The effect of Aβ on PKCε protein expression was assessed with western blot analysis. There was no significant difference on the days 0, 5, 10, and 30 after the injection in the sham and also Aβ-treated groups using one-way ANOVA. Aβ significantly downregulated the PKCε protein expression in the hippocampus of treated groups on day 30 (P<0.05); however, there was no significant difference in PKCε expression in platelet sample (Figure 7 A & B).
4. Discussion
AD is a neurodegenerative disorder that affects several parts of the brain, such as the hippocampus, olfactory bulb, cortical regions, cerebellum, and hypothalamus (El Khoury, Gratuze, Papon, Bretteville, & Planel, 2014). Aβ is one of the most important factors to induce AD. It affects the hippocampus and platelet PKCε-load leading to memory and neurobehavioral profile impairment (Roher et al., 2009).
We aimed at finding the effect of AD on the activity and expression of PKC in reducing spatial learning and memory by examining the hippocampus and platelet of adult rats. The MWM test was employed to study spatial learning and memory. In this regard, traveled distance, escape latencies, and visual platform and probe tests (time spent in the target quadrant) were evaluated. The results showed that the traveled distance was significantly longer and escape latency was significantly shorter in Aβ-treated groups. Aβ has widely been used to create an AD model (Frid, Anisimov, & Popovic, 2007; Hoppe et al., 2013; Majlessi, Choopani, Kamalinejad, & Azizi, 2012; Prakash, Medhi, & Chopra, 2013; White, Manelli, Holmberg, Van Eldik, & Ladu, 2005); therefore, we injected Aβ (1-42) in the rat brain, to simulate a neurodegenerative brain in the present study.
To examine the role of PKC in the pathophysiology of AD, we measured the total PKC activity and also PKC isoenzyme, PKCε, mRNA expression, and protein levels in the platelets and CA1 region of the hippocampus in the treated and sham groups. PKC activity assessment showed that its activity in the AD brain group was lower than the sham group, which was significant 10 days after Aβ injection, and this significant difference was observed 30 days after Aβ injection in platelet samples. The small obtained standard error demonstrated the reliability of this survey on subjects.
The observed reduced activity was consistent with the results of previous studies. Reportedly, the reduced activity of two PKC isoenzymes of α and ε is indirectly associated with enhanced Aβ levels in an AD transgenic mouse model. They also showed that PKC activators could also prohibit the amyloidogenic pathway via inhibition of the β-site of APP cleaving enzyme (Hongpaisan et al., 2011). Other studies also demonstrate that PKCε activity is significantly lower in the AD brain than the control group (Matsushima, Shimohama, Chachin, Taniguchi, & Kimura, 1996; Saitoh et al., 1989). Lanius R.A et al., (1997) showed that there were no significant differences in PKC activity in the frontal, motor, temporal, and parietal cortex or also in the leukocytes and platelets of AD patients and controls. In addition to the changes in PKC activity, we observed some differences in PKCε gene expression and also protein level in the CA1 region of the hippocampus between AD and sham groups.
One month after Aβ injection, the expression ratio of PKCε was significantly changed in the hippocampal tissue compared with the sham groups, but there was no significant difference in its expression in platelets. Unsworth, Smith, Gissen, Watson, & Pears (2011) reported that PKCε negatively regulate ADP-induced aggregation and dense granule secretion. Murugappan et al., study was conducted on platelets containing thromboxane. This lipid generates ADP, which reinforces aggregation and causes dense granule secretion in platelets (Murugappan, Shankar, & Kunapuli, 2004). Also, some studies have demonstrated that aspirin or indomethacin abolished the generation of thromboxane. Under these conditions, loss of PKCε in murine platelets negatively regulates intracellular calcium mobilization, which regulates thromboxane generation (Bynagari-Settipalli et al., 2012).
The insignificant difference in the PKCε expression in platelets may be due to their low level. Buensuceso et al., (Buensuceso et al., 2005) were unable to detect PKCε protein in human platelets and another study attempted to address this through concentration of PKCε by immunoprecipitation, which was unsuccessful, as well. They mentioned that PKCε is expressed at high levels in mouse but not human platelets, and also the differences in batches of antibodies and absence or existence of PKCε may explain the inconsistent results (Pears et al., 2008).
Quantitation of isoform-specific immunoreactivity was assessed by western blot analysis. It showed that AD significantly downregulated the PKCε protein expression in the hippocampus of the treated groups on day 30, but there was no significant difference in PKCε expression in the platelet sample. These results confirmed the expression level of PKCε gene using real-time PCR. These determinations have been done in several previous reports with conflicting results (Bosman, 1992; Lanius et al., 1997; Matsushima et al., 1996; Molchan et al., 1993).
Several studies also have assessed the PKC activity and protein level using quantitative autoradiography or immunological assays, like western blots in AD and sham groups. They did not find a significant difference between AD and sham tissues (Masliah et al., 1990; Shimohama et al., 1993). Western blot analysis was used to measure PKCε and PKCα in hippocampal neurons. For these measurements, the entire hippocampal areas were assessed (Bar-Am, Yogev-Falach, Amit, Sagi, & Youdim, 2004). Based on the reported results, subtle changes in protein levels and or compartments containing protein in distinct areas of the hippocampus were found using immunohistochemistry methods. It has also demonstrated that a decrease in PKCε protein level in the hippocampal CA1 region of Tg2576 mice was due to the decreased number of presynaptic terminals containing PKCε (Hongpaisan, Sun, & Alkon, 2011).
The intracellular production of Aβ can be a potential threat to the cells. The enhancement of Aβ concentration increases the risk of its aggregation and the occurrence of degenerative changes (Oh et al., 2005). These events are observed in transgenic mice and also in areas known to be affected by AD pathology (Chui et al., 1999; Wirths et al., 2001), such as the hippocampus (Gouras et al., 2000). Different isoforms of PKC may be involved and can also play a role in the complex regulation of APP metabolism. Many of these enzymes have been shown defective in AD (Gasparini et al., 1998). In some cases, these defects were associated with aberrant APP metabolism (Bergamaschi et al., 1995; Govoni et al., 1993; Govoni et al., 1996; Vestling et al., 1999).
5. conclusion
In conclusion, the expression ratio and protein expression level of PKCε reduced on day 30 following the injection of Aβ in the hippocampal tissues, but no significant difference between AD and sham groups were observed in platelets that may result from the low concentration of PKCε or insensitive methods to show the expression ratio of the mentioned isoform. Also, in our results, PKC activity in the hippocampus decreased after day 10, but PKCε expression decreased on day 30, suggesting that PKC isozymes other than PKCε may contribute to this change.
Ethical Considerations
Compliance with ethical guidelines
All experiments were executed according to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication; No. 80-23, revised 1996).
Funding
This work was supported by Pasteur Institute of Iran.
Authors' contributions
Investigation: Sara Amiri; Conducted the experiments: Sara Amiri, Baharak Khoshkholgh-Sima; Data analysis: Sara Amiri, Marzieh Dehghan Shasaltaneh; Writing-review & editing: Sara Amiri, Marzieh Dehghan Shasaltaneh; Supervision: Nasser Naghdi, Kayhan Azadmanesh; and All authors have read and approved the manuscript before submission.
Conflict of interest
The authors declared no conflict of interests.
Acknowledgments
We wish to thank Mrs. Zahra Shahosseini for her technical assistance at the Virology Department of Pasteur Institute of Iran.
References
Bergamaschi, S., Binetti, G., Govoni, S., Wetsel, W. C., Battaini, F., Trabucchi, M., et al., (1995). Defective phorbol ester-stimulated secretion of beta-amyloid precursor protein from Alzheimer’s disease fibroblasts. Neuroscience Letters, 201(1), 1-5. [DOI:10.1016/0304-3940(95)12168-4]
Bosman, G. J. C. G. M., Stekhoven, J. S., Melenhorst, J. J., Van Zuylen, A. J., Bartholomeus, I. G. P., Van Kalmthout, P. J. C., et al., (1992). Are thrombocyte membranes altered in Alzheimer’s disease? A morphometric and biochemical study. Neurobiology of Aging, 13(6), 711-6. [DOI:10.1016/0197-4580(92)90094-E]
Buensuceso, C. S., Obergfell, A., Soriani, A., Eto, K., Kiosses, W. B., Arias-Salgado, E. G., et al., (2005). Regulation of outside-in signaling in platelets by integrin-associated protein kinase C beta. Journal of Biological Chemistry, 280(1), 644-53. [DOI:10.1074/jbc.M410229200] [PMID]
Bynagari-Settipalli, Y. S., Lakhani, P., Jin, J., Bhavaraju, K., Rico, M. C., Kim, S., et al., (2012). Protein kinase C isoform epsilon negatively regulates ADP-induced calcium mobilization and thromboxane generation in platelets. Arteriosclerosis, Thrombosis, and Vascular Biology, 32(5), 1211-9. [DOI:10.1161/ATVBAHA.111.242388] [PMID] [PMCID]
Catricala, S., Torti, M., & Ricevuti, G. (2012). Alzheimer disease and platelets: How’s that relevant. Immunity & Ageing, 9(1), 20. [DOI:10.1186/1742-4933-9-20] [PMID] [PMCID]
Chui, D. H., Tanahashi, H., Ozawa, K., Ikeda, S., Checler, F., Ueda, O., et al., (1999). Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nature Medicine, 5(5), 560-4. [DOI:10.1038/8438] [PMID]
De Montigny, A., Elhiri, I., Allyson, J., Cyr, M., & Massicotte, G. (2013). NMDA reduces Tau phosphorylation in rat hippocampal slices by targeting NR2A receptors, GSK3beta, and PKC activities. Neural Plasticity, 2013, 261593. [DOI:10.1155/2013/261593] [PMID] [PMCID]
Deane, R., & Zlokovic, B. V. (2007). Role of the blood-brain barrier in the pathogenesis of Alzheimer’s disease. Current Alzheimer Research, 4(2), 191-7. [DOI:10.2174/156720507780362245] [PMID]
El Khoury, N. B., Gratuze, M., Papon, M. A., Bretteville, A., & Planel, E. (2014). Insulin dysfunction and Tau pathology. Frontiers in Cellular Neuroscience, 8, 22. [DOI:10.3389/fncel.2014.00022] [PMID] [PMCID]
Frid, P., Anisimov, S. V., & Popovic, N. (2007). Congo red and protein aggregation in neurodegenerative diseases. Brain Research Reviews, 53(1), 135-60. [DOI:10.1016/j.brainresrev.2006.08.001] [PMID]
Gasparini, L., Racchi, M., Binetti, G., Trabucchi, M., Solerte, S. B., Alkon, D., et al., (1998). Peripheral markers in testing pathophysiological hypotheses and diagnosing Alzheimer’s disease. The FASEB Journal, 12(1), 17-34. [DOI:10.1096/fasebj.12.1.17] [PMID]
Gouras, G. K., Tsai, J., Naslund, J., Vincent, B., Edgar, M., Checler, F., et al., (2000). Intraneuronal Abeta42 accumulation in human brain. The American Journal of Pathology, 156(1), 15-20. [DOI:10.1016/S0002-9440(10)64700-1]
Govoni, S., Bergamaschi, S., Racchi, M., Battaini, F., Binetti, G., Bianchetti, A., et al., (1993). Cytosol protein kinase C downregulation in fibroblasts from Alzheimer’s disease patients. Neurology, 43(12), 2581-6. [DOI:10.1212/WNL.43.12.2581] [PMID]
Govoni, S., Racchi, M., Bergamaschi, S., Trabucchi, M., Battaini, F., Bianchetti, A., et al., (1996). Defective protein kinase C alpha leads to impaired secretion of soluble beta-amyloid precursor protein from Alzheimer’s disease fibroblasts. Annals of the New York Academy of Sciences, 777, 332-7. [DOI:10.1111/j.1749-6632.1996.tb34442.x] [PMID]
Hong-Qi, Y., Zhi-Kun, S., & Sheng-Di, C. (2012). Current advances in the treatment of Alzheimer’s disease: Focused on considerations targeting Abeta and tau. Translational Neurodegeneration, 1(1), 21. [DOI:10.1186/2047-9158-1-21] [PMID] [PMCID]
Hongpaisan, J., Sun, M. K., & Alkon, D. L. (2011). PKC epsilon activation prevents synaptic loss, Abeta elevation, and cognitive deficits in Alzheimer’s disease transgenic mice. The Journal of Neuroscience, 31(2), 630-43. [DOI:10.1523/JNEUROSCI.5209-10.2011] [PMID] [PMCID]
Hoppe, J. B., Coradini, K., Frozza, R. L., Oliveira, C. M., Meneghetti, A. B., Bernardi, A., et al., (2013). Free and nanoencapsulated curcumin suppress beta-amyloid-induced cognitive impairments in rats: Involvement of BDNF and Akt/GSK-3beta signaling pathway. Neurobiology of Learning and Memory, 106, 134-44. [DOI:10.1016/j.nlm.2013.08.001] [PMID]
Huang, Y., & Mucke, L. (2012). Alzheimer mechanisms and therapeutic strategies. Cell, 148(6), 1204-22. [DOI:10.1016/j.cell.2012.02.040] [PMID] [PMCID]
Humpel, C. (2011). Identifying and validating biomarkers for Alzheimer’s disease. Trends in Biotechnology, 29(1), 26-32. [DOI:10.1016/j.tibtech.2010.09.007] [PMID] [PMCID]
Jaremo, P., Milovanovic, M., Buller, C., Nilsson, S., & Winblad, B. (2012). Low-density platelet populations demonstrate low in vivo activity in sporadic Alzheimer disease. Platelets, 23(2), 116-20. [DOI:10.3109/09537104.2011.593654] [PMID]
Khan, T. K., & Alkon, D. L. (2010). Early diagnostic accuracy and pathophysiologic relevance of an autopsy-confirmed Alzheimer’s disease peripheral biomarker. Neurobiology of Aging, 31(6), 889-900. [DOI:10.1016/j.neurobiolaging.2008.07.010] [PMID]
Kumar, A., Singh, A., & Ekavali, E. (2015). A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacological Reports, 67(2), 195-203. [DOI:10.1016/j.pharep.2014.09.004] [PMID]
Lanius, R. A., Wagey, R., Sahl, B., Beattie, B. L., Feldman, H., Pelech, S. L., et al., (1997). Protein kinase C activity and protein levels in Alzheimer’s disease. Brain Research, 764(1-2), 75-80. [DOI:10.1016/S0006-8993(97)00422-8]
Lucke-Wold, B. P., Turner, R. C., Logsdon, A. F., Simpkins, J. W., Alkon, D. L., Smith, K. E., et al., (2015). Common mechanisms of Alzheimer’s disease and ischemic stroke: The role of protein kinase C in the progression of age-related neurodegeneration. Journal of Alzheimer’s Disease, 43(3), 711-24. [DOI:10.3233/JAD-141422] [PMID] [PMCID]
Majlessi, N., Choopani, S., Kamalinejad, M., & Azizi, Z. (2012). Amelioration of amyloid beta-induced cognitive deficits by Zataria multiflora Boiss: Essential oil in a rat model of Alzheimer’s disease. CNS Neuroscience & Therapeutics, 18(4), 295-301. [DOI:10.1111/j.1755-5949.2011.00237.x] [PMID] [PMCID]
Masliah, E., Cole, G., Shimohama, S., Hansen, L., DeTeresa, R., Terry, R. D., et al., (1990). Differential involvement of protein kinase C isozymes in Alzheimer’s disease. The Journal of Neuroscience, 10(7), 2113-24. [DOI:10.1523/JNEUROSCI.10-07-02113.1990] [PMID] [PMCID]
Matsushima, H., Shimohama, S., Chachin, M., Taniguchi, T., & Kimura, J. (1996). Ca2+-dependent and Ca2+-independent protein kinase C changes in the brain of patients with Alzheimer’s disease. Journal of Neurochemistry, 67(1), 317-23. [DOI:10.1046/j.1471-4159.1996.67010317.x] [PMID]
Matsushima, H., Shimohama, S., Tanaka, S., Taniguchi, T., Hagiwara, M., Hidaka, H., et al., (1994). Platelet protein kinase C levels in Alzheimer’s disease. Neurobiology of Aging, 15(6), 671-4. [DOI:10.1016/0197-4580(94)90047-7]
Milovanovic, M., Eriksson, K., Winblad, B., Nilsson, S., Lindahl, T. L., Post, C., et al., (2014). Alzheimer and platelets: Low-density platelet populations reveal increased serotonin content in Alzheimer type dementia. Clinical Biochemistry, 47(15), 51-3. [DOI:10.1016/j.clinbiochem.2014.07.007] [PMID]
Molchan, S. E., Manji, H., Chen, G., Dou, L., Little, J., Potter, W. Z., et al., (1993). Effects of chronic lithium treatment on platelet PKC isozymes in Alzheimer’s and elderly control subjects. Neuroscience Letters, 162(1-2), 187-91. [DOI:10.1016/0304-3940(93)90592-9]
Murugappan, S., Shankar, H., & Kunapuli, S. P. (2004). Platelet receptors for adenine nucleotides and thromboxane A2. Seminars in Thrombosis and Hemostasis, 30(4), 411-8. [DOI:10.1055/s-2004-833476] [PMID]
Nelson, T. J., Sun, M. K., Hongpaisan, J., & Alkon, D. L. (2008). Insulin, PKC signaling pathways and synaptic remodeling during memory storage and neuronal repair. European Journal of Pharmacology, 585(1), 76-87. [DOI:10.1016/j.ejphar.2008.01.051] [PMID]
Bar-Am O, Yogev-Falach M, Amit T, Sagi Y, Youdim MB. Youdim, Regulation of protein kinase C by the anti-Parkinson drug, MAO-B inhibitor, rasagiline and its derivatives, in vivo. Journal of neurochemistry, 89(5), 1119-25.
Oh, S., Hong, H. S., Hwang, E., Sim, H. J., Lee, W., Shin, S. J., et al., (2005). Amyloid peptide attenuates the proteasome activity in neuronal cells. Mechanisms of Ageing and Development, 126(12), 1292-9. [DOI:10.1016/j.mad.2005.07.006] [PMID]
Pacheco-Quinto, J., & Eckman, E. A. (2013). Endothelin-converting enzymes degrade intracellular beta-amyloid produced within the endosomal/lysosomal pathway and autophagosomes. Journal of Biological Chemistry, 288(8), 5606-15. [DOI:10.1074/jbc.M112.422964] [PMID] [PMCID]
Paxinos., G., & Watson., C. (1986). The rat brain in stereotaxic coordinates (2nd ed.). Orlando, FL: Academic Press.
Pears, C. J., Thornber, K., Auger, J. M., Hughes, C. E., Grygielska, B., Protty, M. B., et al., (2008). Differential roles of the PKC novel isoforms, PKCdelta and PKCepsilon, in mouse and human platelets. PLoS One, 3(11), e3793. [DOI:10.1371/journal.pone.0003793] [PMID] [PMCID]
Prakash, A., Medhi, B., & Chopra, K. (2013). Granulocyte Colony Stimulating Factor (GCSF) improves memory and neurobehavior in an amyloid-beta induced experimental model of Alzheimer’s disease. Pharmacology Biochemistry and Behavior, 110, 46-57. [DOI:10.1016/j.pbb.2013.05.015] [PMID]
Roher, A. E., Esh, C. L., Kokjohn, T. A., Castano, E. M., Van Vickle, G. D., Kalback, W. M., et al., (2009). Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer’s disease. Alzheimer’s & Dementia, 5(1), 18-29. [DOI:10.1016/j.jalz.2008.10.004] [PMID] [PMCID]
Saitoh, T., Sundsmo, M., Roch, J. M., Kimura, N., Cole, G., Schubert, D., et al., (1989). Secreted form of amyloid beta protein precursor is involved in the growth regulation of fibroblasts. Cell, 58(4), 615-22. [DOI:10.1016/0092-8674(89)90096-2]
Shimohama S, N. M., Matsushima H, Kimura J, Kameyama M, Hagiwara M, Hidaka H, et al., (1993). Assessment of protein kinase C isozymes by two-site enzyme immunoassay in human brains and changes in Alzheimer’s disease. Neurology, 43(7), 1407-13. [DOI:10.1212/WNL.43.7.1407] [PMID]
Sun, M. K., & Alkon, D. L. (2014). The “memory kinases”: Roles of PKC isoforms in signal processing and memory formation. Progress in Molecular Biology and Translational Science, 122, 31-59. [DOI:10.1016/B978-0-12-420170-5.00002-7] [PMID]
Sun, M. K., Nelson, T. J., & Alkon, D. L. (2015). Towards universal therapeutics for memory disorders. Trends in Pharmacological Sciences, 36(6), 384-94. [DOI:10.1016/j.tips.2015.04.004] [PMID]
Unsworth, A. J., Smith, H., Gissen, P., Watson, S. P., & Pears, C. J. (2011). Submaximal inhibition of protein kinase C restores ADP-induced dense granule secretion in platelets in the presence of Ca2+. Journal of Biological Chemistry, 286(24), 21073-82. [DOI:10.1074/jbc.M110.187138] [PMID] [PMCID]
Vestling, M., Cedazo-Minguez, A., Adem, A., Wiehager, B., Racchi, M., Lannfelt, L., et al., (1999). Protein kinase C and amyloid precursor protein processing in skin fibroblasts from sporadic and familial Alzheimer’s disease cases. Biochimica et Biophysica Acta, 1453(3), 341-50. [DOI:10.1016/S0925-4439(99)00003-4]
Wang, H. Y., Pisano, M. R., & Friedman, E. (1994). Attenuated protein kinase C activity and translocation in Alzheimer’s disease brain. Neurobiology of Aging, 15(3), 293-8. [DOI:10.1016/0197-4580(94)90023-X]
White, J. A., Manelli, A. M., Holmberg, K. H., Van Eldik, L. J., & Ladu, M. J. (2005). Differential effects of oligomeric and fibrillar amyloid-beta 1-42 on astrocyte-mediated inflammation. Neurobiology of Disease, 18(3), 459-65. [DOI:10.1016/j.nbd.2004.12.013] [PMID]
Wirths, O., Multhaup, G., Czech, C., Blanchard, V., Moussaoui, S., Tremp, G., et al., (2001). Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neuroscience Letters, 306(1-2), 116-20. [DOI:10.1016/S0304-3940(01)01876-6]
AD is a neurodegenerative disorder that affects several parts of the brain, such as the hippocampus, olfactory bulb, cortical regions, cerebellum, and hypothalamus (El Khoury, Gratuze, Papon, Bretteville, & Planel, 2014). Aβ is one of the most important factors to induce AD. It affects the hippocampus and platelet PKCε-load leading to memory and neurobehavioral profile impairment (Roher et al., 2009).
We aimed at finding the effect of AD on the activity and expression of PKC in reducing spatial learning and memory by examining the hippocampus and platelet of adult rats. The MWM test was employed to study spatial learning and memory. In this regard, traveled distance, escape latencies, and visual platform and probe tests (time spent in the target quadrant) were evaluated. The results showed that the traveled distance was significantly longer and escape latency was significantly shorter in Aβ-treated groups. Aβ has widely been used to create an AD model (Frid, Anisimov, & Popovic, 2007; Hoppe et al., 2013; Majlessi, Choopani, Kamalinejad, & Azizi, 2012; Prakash, Medhi, & Chopra, 2013; White, Manelli, Holmberg, Van Eldik, & Ladu, 2005); therefore, we injected Aβ (1-42) in the rat brain, to simulate a neurodegenerative brain in the present study.
To examine the role of PKC in the pathophysiology of AD, we measured the total PKC activity and also PKC isoenzyme, PKCε, mRNA expression, and protein levels in the platelets and CA1 region of the hippocampus in the treated and sham groups. PKC activity assessment showed that its activity in the AD brain group was lower than the sham group, which was significant 10 days after Aβ injection, and this significant difference was observed 30 days after Aβ injection in platelet samples. The small obtained standard error demonstrated the reliability of this survey on subjects.
The observed reduced activity was consistent with the results of previous studies. Reportedly, the reduced activity of two PKC isoenzymes of α and ε is indirectly associated with enhanced Aβ levels in an AD transgenic mouse model. They also showed that PKC activators could also prohibit the amyloidogenic pathway via inhibition of the β-site of APP cleaving enzyme (Hongpaisan et al., 2011). Other studies also demonstrate that PKCε activity is significantly lower in the AD brain than the control group (Matsushima, Shimohama, Chachin, Taniguchi, & Kimura, 1996; Saitoh et al., 1989). Lanius R.A et al., (1997) showed that there were no significant differences in PKC activity in the frontal, motor, temporal, and parietal cortex or also in the leukocytes and platelets of AD patients and controls. In addition to the changes in PKC activity, we observed some differences in PKCε gene expression and also protein level in the CA1 region of the hippocampus between AD and sham groups.
One month after Aβ injection, the expression ratio of PKCε was significantly changed in the hippocampal tissue compared with the sham groups, but there was no significant difference in its expression in platelets. Unsworth, Smith, Gissen, Watson, & Pears (2011) reported that PKCε negatively regulate ADP-induced aggregation and dense granule secretion. Murugappan et al., study was conducted on platelets containing thromboxane. This lipid generates ADP, which reinforces aggregation and causes dense granule secretion in platelets (Murugappan, Shankar, & Kunapuli, 2004). Also, some studies have demonstrated that aspirin or indomethacin abolished the generation of thromboxane. Under these conditions, loss of PKCε in murine platelets negatively regulates intracellular calcium mobilization, which regulates thromboxane generation (Bynagari-Settipalli et al., 2012).
The insignificant difference in the PKCε expression in platelets may be due to their low level. Buensuceso et al., (Buensuceso et al., 2005) were unable to detect PKCε protein in human platelets and another study attempted to address this through concentration of PKCε by immunoprecipitation, which was unsuccessful, as well. They mentioned that PKCε is expressed at high levels in mouse but not human platelets, and also the differences in batches of antibodies and absence or existence of PKCε may explain the inconsistent results (Pears et al., 2008).
Quantitation of isoform-specific immunoreactivity was assessed by western blot analysis. It showed that AD significantly downregulated the PKCε protein expression in the hippocampus of the treated groups on day 30, but there was no significant difference in PKCε expression in the platelet sample. These results confirmed the expression level of PKCε gene using real-time PCR. These determinations have been done in several previous reports with conflicting results (Bosman, 1992; Lanius et al., 1997; Matsushima et al., 1996; Molchan et al., 1993).
Several studies also have assessed the PKC activity and protein level using quantitative autoradiography or immunological assays, like western blots in AD and sham groups. They did not find a significant difference between AD and sham tissues (Masliah et al., 1990; Shimohama et al., 1993). Western blot analysis was used to measure PKCε and PKCα in hippocampal neurons. For these measurements, the entire hippocampal areas were assessed (Bar-Am, Yogev-Falach, Amit, Sagi, & Youdim, 2004). Based on the reported results, subtle changes in protein levels and or compartments containing protein in distinct areas of the hippocampus were found using immunohistochemistry methods. It has also demonstrated that a decrease in PKCε protein level in the hippocampal CA1 region of Tg2576 mice was due to the decreased number of presynaptic terminals containing PKCε (Hongpaisan, Sun, & Alkon, 2011).
The intracellular production of Aβ can be a potential threat to the cells. The enhancement of Aβ concentration increases the risk of its aggregation and the occurrence of degenerative changes (Oh et al., 2005). These events are observed in transgenic mice and also in areas known to be affected by AD pathology (Chui et al., 1999; Wirths et al., 2001), such as the hippocampus (Gouras et al., 2000). Different isoforms of PKC may be involved and can also play a role in the complex regulation of APP metabolism. Many of these enzymes have been shown defective in AD (Gasparini et al., 1998). In some cases, these defects were associated with aberrant APP metabolism (Bergamaschi et al., 1995; Govoni et al., 1993; Govoni et al., 1996; Vestling et al., 1999).
5. conclusion
In conclusion, the expression ratio and protein expression level of PKCε reduced on day 30 following the injection of Aβ in the hippocampal tissues, but no significant difference between AD and sham groups were observed in platelets that may result from the low concentration of PKCε or insensitive methods to show the expression ratio of the mentioned isoform. Also, in our results, PKC activity in the hippocampus decreased after day 10, but PKCε expression decreased on day 30, suggesting that PKC isozymes other than PKCε may contribute to this change.
Ethical Considerations
Compliance with ethical guidelines
All experiments were executed according to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication; No. 80-23, revised 1996).
Funding
This work was supported by Pasteur Institute of Iran.
Authors' contributions
Investigation: Sara Amiri; Conducted the experiments: Sara Amiri, Baharak Khoshkholgh-Sima; Data analysis: Sara Amiri, Marzieh Dehghan Shasaltaneh; Writing-review & editing: Sara Amiri, Marzieh Dehghan Shasaltaneh; Supervision: Nasser Naghdi, Kayhan Azadmanesh; and All authors have read and approved the manuscript before submission.
Conflict of interest
The authors declared no conflict of interests.
Acknowledgments
We wish to thank Mrs. Zahra Shahosseini for her technical assistance at the Virology Department of Pasteur Institute of Iran.
References
Bergamaschi, S., Binetti, G., Govoni, S., Wetsel, W. C., Battaini, F., Trabucchi, M., et al., (1995). Defective phorbol ester-stimulated secretion of beta-amyloid precursor protein from Alzheimer’s disease fibroblasts. Neuroscience Letters, 201(1), 1-5. [DOI:10.1016/0304-3940(95)12168-4]
Bosman, G. J. C. G. M., Stekhoven, J. S., Melenhorst, J. J., Van Zuylen, A. J., Bartholomeus, I. G. P., Van Kalmthout, P. J. C., et al., (1992). Are thrombocyte membranes altered in Alzheimer’s disease? A morphometric and biochemical study. Neurobiology of Aging, 13(6), 711-6. [DOI:10.1016/0197-4580(92)90094-E]
Buensuceso, C. S., Obergfell, A., Soriani, A., Eto, K., Kiosses, W. B., Arias-Salgado, E. G., et al., (2005). Regulation of outside-in signaling in platelets by integrin-associated protein kinase C beta. Journal of Biological Chemistry, 280(1), 644-53. [DOI:10.1074/jbc.M410229200] [PMID]
Bynagari-Settipalli, Y. S., Lakhani, P., Jin, J., Bhavaraju, K., Rico, M. C., Kim, S., et al., (2012). Protein kinase C isoform epsilon negatively regulates ADP-induced calcium mobilization and thromboxane generation in platelets. Arteriosclerosis, Thrombosis, and Vascular Biology, 32(5), 1211-9. [DOI:10.1161/ATVBAHA.111.242388] [PMID] [PMCID]
Catricala, S., Torti, M., & Ricevuti, G. (2012). Alzheimer disease and platelets: How’s that relevant. Immunity & Ageing, 9(1), 20. [DOI:10.1186/1742-4933-9-20] [PMID] [PMCID]
Chui, D. H., Tanahashi, H., Ozawa, K., Ikeda, S., Checler, F., Ueda, O., et al., (1999). Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nature Medicine, 5(5), 560-4. [DOI:10.1038/8438] [PMID]
De Montigny, A., Elhiri, I., Allyson, J., Cyr, M., & Massicotte, G. (2013). NMDA reduces Tau phosphorylation in rat hippocampal slices by targeting NR2A receptors, GSK3beta, and PKC activities. Neural Plasticity, 2013, 261593. [DOI:10.1155/2013/261593] [PMID] [PMCID]
Deane, R., & Zlokovic, B. V. (2007). Role of the blood-brain barrier in the pathogenesis of Alzheimer’s disease. Current Alzheimer Research, 4(2), 191-7. [DOI:10.2174/156720507780362245] [PMID]
El Khoury, N. B., Gratuze, M., Papon, M. A., Bretteville, A., & Planel, E. (2014). Insulin dysfunction and Tau pathology. Frontiers in Cellular Neuroscience, 8, 22. [DOI:10.3389/fncel.2014.00022] [PMID] [PMCID]
Frid, P., Anisimov, S. V., & Popovic, N. (2007). Congo red and protein aggregation in neurodegenerative diseases. Brain Research Reviews, 53(1), 135-60. [DOI:10.1016/j.brainresrev.2006.08.001] [PMID]
Gasparini, L., Racchi, M., Binetti, G., Trabucchi, M., Solerte, S. B., Alkon, D., et al., (1998). Peripheral markers in testing pathophysiological hypotheses and diagnosing Alzheimer’s disease. The FASEB Journal, 12(1), 17-34. [DOI:10.1096/fasebj.12.1.17] [PMID]
Gouras, G. K., Tsai, J., Naslund, J., Vincent, B., Edgar, M., Checler, F., et al., (2000). Intraneuronal Abeta42 accumulation in human brain. The American Journal of Pathology, 156(1), 15-20. [DOI:10.1016/S0002-9440(10)64700-1]
Govoni, S., Bergamaschi, S., Racchi, M., Battaini, F., Binetti, G., Bianchetti, A., et al., (1993). Cytosol protein kinase C downregulation in fibroblasts from Alzheimer’s disease patients. Neurology, 43(12), 2581-6. [DOI:10.1212/WNL.43.12.2581] [PMID]
Govoni, S., Racchi, M., Bergamaschi, S., Trabucchi, M., Battaini, F., Bianchetti, A., et al., (1996). Defective protein kinase C alpha leads to impaired secretion of soluble beta-amyloid precursor protein from Alzheimer’s disease fibroblasts. Annals of the New York Academy of Sciences, 777, 332-7. [DOI:10.1111/j.1749-6632.1996.tb34442.x] [PMID]
Hong-Qi, Y., Zhi-Kun, S., & Sheng-Di, C. (2012). Current advances in the treatment of Alzheimer’s disease: Focused on considerations targeting Abeta and tau. Translational Neurodegeneration, 1(1), 21. [DOI:10.1186/2047-9158-1-21] [PMID] [PMCID]
Hongpaisan, J., Sun, M. K., & Alkon, D. L. (2011). PKC epsilon activation prevents synaptic loss, Abeta elevation, and cognitive deficits in Alzheimer’s disease transgenic mice. The Journal of Neuroscience, 31(2), 630-43. [DOI:10.1523/JNEUROSCI.5209-10.2011] [PMID] [PMCID]
Hoppe, J. B., Coradini, K., Frozza, R. L., Oliveira, C. M., Meneghetti, A. B., Bernardi, A., et al., (2013). Free and nanoencapsulated curcumin suppress beta-amyloid-induced cognitive impairments in rats: Involvement of BDNF and Akt/GSK-3beta signaling pathway. Neurobiology of Learning and Memory, 106, 134-44. [DOI:10.1016/j.nlm.2013.08.001] [PMID]
Huang, Y., & Mucke, L. (2012). Alzheimer mechanisms and therapeutic strategies. Cell, 148(6), 1204-22. [DOI:10.1016/j.cell.2012.02.040] [PMID] [PMCID]
Humpel, C. (2011). Identifying and validating biomarkers for Alzheimer’s disease. Trends in Biotechnology, 29(1), 26-32. [DOI:10.1016/j.tibtech.2010.09.007] [PMID] [PMCID]
Jaremo, P., Milovanovic, M., Buller, C., Nilsson, S., & Winblad, B. (2012). Low-density platelet populations demonstrate low in vivo activity in sporadic Alzheimer disease. Platelets, 23(2), 116-20. [DOI:10.3109/09537104.2011.593654] [PMID]
Khan, T. K., & Alkon, D. L. (2010). Early diagnostic accuracy and pathophysiologic relevance of an autopsy-confirmed Alzheimer’s disease peripheral biomarker. Neurobiology of Aging, 31(6), 889-900. [DOI:10.1016/j.neurobiolaging.2008.07.010] [PMID]
Kumar, A., Singh, A., & Ekavali, E. (2015). A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacological Reports, 67(2), 195-203. [DOI:10.1016/j.pharep.2014.09.004] [PMID]
Lanius, R. A., Wagey, R., Sahl, B., Beattie, B. L., Feldman, H., Pelech, S. L., et al., (1997). Protein kinase C activity and protein levels in Alzheimer’s disease. Brain Research, 764(1-2), 75-80. [DOI:10.1016/S0006-8993(97)00422-8]
Lucke-Wold, B. P., Turner, R. C., Logsdon, A. F., Simpkins, J. W., Alkon, D. L., Smith, K. E., et al., (2015). Common mechanisms of Alzheimer’s disease and ischemic stroke: The role of protein kinase C in the progression of age-related neurodegeneration. Journal of Alzheimer’s Disease, 43(3), 711-24. [DOI:10.3233/JAD-141422] [PMID] [PMCID]
Majlessi, N., Choopani, S., Kamalinejad, M., & Azizi, Z. (2012). Amelioration of amyloid beta-induced cognitive deficits by Zataria multiflora Boiss: Essential oil in a rat model of Alzheimer’s disease. CNS Neuroscience & Therapeutics, 18(4), 295-301. [DOI:10.1111/j.1755-5949.2011.00237.x] [PMID] [PMCID]
Masliah, E., Cole, G., Shimohama, S., Hansen, L., DeTeresa, R., Terry, R. D., et al., (1990). Differential involvement of protein kinase C isozymes in Alzheimer’s disease. The Journal of Neuroscience, 10(7), 2113-24. [DOI:10.1523/JNEUROSCI.10-07-02113.1990] [PMID] [PMCID]
Matsushima, H., Shimohama, S., Chachin, M., Taniguchi, T., & Kimura, J. (1996). Ca2+-dependent and Ca2+-independent protein kinase C changes in the brain of patients with Alzheimer’s disease. Journal of Neurochemistry, 67(1), 317-23. [DOI:10.1046/j.1471-4159.1996.67010317.x] [PMID]
Matsushima, H., Shimohama, S., Tanaka, S., Taniguchi, T., Hagiwara, M., Hidaka, H., et al., (1994). Platelet protein kinase C levels in Alzheimer’s disease. Neurobiology of Aging, 15(6), 671-4. [DOI:10.1016/0197-4580(94)90047-7]
Milovanovic, M., Eriksson, K., Winblad, B., Nilsson, S., Lindahl, T. L., Post, C., et al., (2014). Alzheimer and platelets: Low-density platelet populations reveal increased serotonin content in Alzheimer type dementia. Clinical Biochemistry, 47(15), 51-3. [DOI:10.1016/j.clinbiochem.2014.07.007] [PMID]
Molchan, S. E., Manji, H., Chen, G., Dou, L., Little, J., Potter, W. Z., et al., (1993). Effects of chronic lithium treatment on platelet PKC isozymes in Alzheimer’s and elderly control subjects. Neuroscience Letters, 162(1-2), 187-91. [DOI:10.1016/0304-3940(93)90592-9]
Murugappan, S., Shankar, H., & Kunapuli, S. P. (2004). Platelet receptors for adenine nucleotides and thromboxane A2. Seminars in Thrombosis and Hemostasis, 30(4), 411-8. [DOI:10.1055/s-2004-833476] [PMID]
Nelson, T. J., Sun, M. K., Hongpaisan, J., & Alkon, D. L. (2008). Insulin, PKC signaling pathways and synaptic remodeling during memory storage and neuronal repair. European Journal of Pharmacology, 585(1), 76-87. [DOI:10.1016/j.ejphar.2008.01.051] [PMID]
Bar-Am O, Yogev-Falach M, Amit T, Sagi Y, Youdim MB. Youdim, Regulation of protein kinase C by the anti-Parkinson drug, MAO-B inhibitor, rasagiline and its derivatives, in vivo. Journal of neurochemistry, 89(5), 1119-25.
Oh, S., Hong, H. S., Hwang, E., Sim, H. J., Lee, W., Shin, S. J., et al., (2005). Amyloid peptide attenuates the proteasome activity in neuronal cells. Mechanisms of Ageing and Development, 126(12), 1292-9. [DOI:10.1016/j.mad.2005.07.006] [PMID]
Pacheco-Quinto, J., & Eckman, E. A. (2013). Endothelin-converting enzymes degrade intracellular beta-amyloid produced within the endosomal/lysosomal pathway and autophagosomes. Journal of Biological Chemistry, 288(8), 5606-15. [DOI:10.1074/jbc.M112.422964] [PMID] [PMCID]
Paxinos., G., & Watson., C. (1986). The rat brain in stereotaxic coordinates (2nd ed.). Orlando, FL: Academic Press.
Pears, C. J., Thornber, K., Auger, J. M., Hughes, C. E., Grygielska, B., Protty, M. B., et al., (2008). Differential roles of the PKC novel isoforms, PKCdelta and PKCepsilon, in mouse and human platelets. PLoS One, 3(11), e3793. [DOI:10.1371/journal.pone.0003793] [PMID] [PMCID]
Prakash, A., Medhi, B., & Chopra, K. (2013). Granulocyte Colony Stimulating Factor (GCSF) improves memory and neurobehavior in an amyloid-beta induced experimental model of Alzheimer’s disease. Pharmacology Biochemistry and Behavior, 110, 46-57. [DOI:10.1016/j.pbb.2013.05.015] [PMID]
Roher, A. E., Esh, C. L., Kokjohn, T. A., Castano, E. M., Van Vickle, G. D., Kalback, W. M., et al., (2009). Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer’s disease. Alzheimer’s & Dementia, 5(1), 18-29. [DOI:10.1016/j.jalz.2008.10.004] [PMID] [PMCID]
Saitoh, T., Sundsmo, M., Roch, J. M., Kimura, N., Cole, G., Schubert, D., et al., (1989). Secreted form of amyloid beta protein precursor is involved in the growth regulation of fibroblasts. Cell, 58(4), 615-22. [DOI:10.1016/0092-8674(89)90096-2]
Shimohama S, N. M., Matsushima H, Kimura J, Kameyama M, Hagiwara M, Hidaka H, et al., (1993). Assessment of protein kinase C isozymes by two-site enzyme immunoassay in human brains and changes in Alzheimer’s disease. Neurology, 43(7), 1407-13. [DOI:10.1212/WNL.43.7.1407] [PMID]
Sun, M. K., & Alkon, D. L. (2014). The “memory kinases”: Roles of PKC isoforms in signal processing and memory formation. Progress in Molecular Biology and Translational Science, 122, 31-59. [DOI:10.1016/B978-0-12-420170-5.00002-7] [PMID]
Sun, M. K., Nelson, T. J., & Alkon, D. L. (2015). Towards universal therapeutics for memory disorders. Trends in Pharmacological Sciences, 36(6), 384-94. [DOI:10.1016/j.tips.2015.04.004] [PMID]
Unsworth, A. J., Smith, H., Gissen, P., Watson, S. P., & Pears, C. J. (2011). Submaximal inhibition of protein kinase C restores ADP-induced dense granule secretion in platelets in the presence of Ca2+. Journal of Biological Chemistry, 286(24), 21073-82. [DOI:10.1074/jbc.M110.187138] [PMID] [PMCID]
Vestling, M., Cedazo-Minguez, A., Adem, A., Wiehager, B., Racchi, M., Lannfelt, L., et al., (1999). Protein kinase C and amyloid precursor protein processing in skin fibroblasts from sporadic and familial Alzheimer’s disease cases. Biochimica et Biophysica Acta, 1453(3), 341-50. [DOI:10.1016/S0925-4439(99)00003-4]
Wang, H. Y., Pisano, M. R., & Friedman, E. (1994). Attenuated protein kinase C activity and translocation in Alzheimer’s disease brain. Neurobiology of Aging, 15(3), 293-8. [DOI:10.1016/0197-4580(94)90023-X]
White, J. A., Manelli, A. M., Holmberg, K. H., Van Eldik, L. J., & Ladu, M. J. (2005). Differential effects of oligomeric and fibrillar amyloid-beta 1-42 on astrocyte-mediated inflammation. Neurobiology of Disease, 18(3), 459-65. [DOI:10.1016/j.nbd.2004.12.013] [PMID]
Wirths, O., Multhaup, G., Czech, C., Blanchard, V., Moussaoui, S., Tremp, G., et al., (2001). Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neuroscience Letters, 306(1-2), 116-20. [DOI:10.1016/S0304-3940(01)01876-6]
Type of Study: Original |
Subject:
Behavioral Neuroscience
Received: 2018/12/3 | Accepted: 2019/02/7 | Published: 2019/11/1
Received: 2018/12/3 | Accepted: 2019/02/7 | Published: 2019/11/1
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
