

Volume 8, Issue 3 (May & June 2017 -- 2017)
BCN 2017, 8(3): 173-182 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Golabi S, Zaringhalam J, Manaheji H. Research Paper: The Impact of Synovial NF-ĸB Activation on Apoptosis Pattern Change During Adjuvant-induced Inflammation. BCN 2017; 8 (3) :173-182
URL: http://bcn.iums.ac.ir/article-1-801-en.html
URL: http://bcn.iums.ac.ir/article-1-801-en.html


1- Neurophysiology Research Center, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
Full-Text [PDF 725 kb]
| Abstract (HTML)
Full-Text:
1. Introduction
Inflammation is the primary and, essentially, important physiological reaction that normally resolves with the restoration of normal structure and function of tissue. It has symptoms such as edema, warmth, and redness (Akhtari, Zaringhalam, Eidi, Manaheji, & Tekieh, 2012; Foyet et al., 2014). Chronic inflammation is a long-duration inflammation, which can cause tissue damage and loss of function (Akhtari, et al,. 2012; Multag, 2012). Chronic inflammation is also along with increased local and systemic levels of certain inflammatory mediators such as cytokines and chemokines (Zhang, Yin, Xu, Wu, & Chen, 2012; Zaringhalam, Tekieh, Manaheji, & Akhtari, 2013). Rheumatoid Arthritis (RA) is a systemic, chronic inflammatory disorder that affects approximately 1% of the universal population. RA is described by symptoms such as joint swelling, pain, and inflammatory synovitis, which leads to hyperplasia (Kannan, Ortmann, & Kimpel, 2005; Yoshida, Ochiai, Matsuno, Panayi, & Corrigall, 2011; Foyet et al., 2014). Complete Freund’s Adjuvant (CFA)-induced arthritis is an inflammatory model commonly recommended for acute and chronic animal inflammation model (Rezazadeh, Zaringhalam, Manaheji, & Kebryaeezadeh, 2009; Zaringhalam, et al., 2013). This model is valuable to evaluate various behavioral and molecular changes made during acute and chronic inflammation due to its similarity to human RA (Taniguchi, Kanai, Kawamoto, Endo, & Higashino, 2004; Zaringhalam, et al., 2013).
Previous studies demonstrated that apoptosis dysregulation is the main pathophysiological mechanism by which mild prime inflammatory diseases progress to sever chronic inflammatory disorders (Anderson, 1996; Hayashi, 2003). Some studies demonstrated that apoptosis or programmed cell death is a central check-point, which limits the intensity of immune response (Anderson, 1996). Immune system activity and homeostasis are strongly related to apoptosis and chronic inflammation, which can be associated with apoptosis failure (Vandivier, Henson, & Douglas, 2006; Favaloro, Allocati, Graziano, Di Ilio, & De Laurenzi, 2012).
In this regard, studies showed that apoptosis impairment had an important and determinant role in the pathogenesis of lung chronic inflammatory diseases such as Acute Respiratory Distress Syndrome (ARDS), community-acquired pneumonia, Chronic Obstructive Pulmonary Disease (COPD), and Cystic Fibrosis (CF); therefore, in ARDS and community-acquired pneumonia, the number of observed apoptotic cells remain quite low, and in patients with COPD and CF apoptotic cells are increased (Vandivier et al., 2006). Moreover, studies pointed that apoptosis had an important and pivotal role in RA pathogenesis. In this regard, it was revealed that during RA, the synovial fibroblasts were resistant to apoptosis and their resistance correlated with the disease progression. Several anti-apoptotic molecules such as Sentrin‐1/small Ubiquitin‐like Modifier (SUMO)‐1, downstream modulators of Fas signaling and transcriptional regulators such as NF-κB, Stat3, and p53 are nominated to modulate apoptosis in this situation (Baier, Meineckel, Gay, & Pap, 2003; Vandivier et al, 2006; Favaloro et al., 2012; Foyet et al., 2014). Emerging body of proofs suggests that the reconnaissance of better approaches to induce selective apoptosis may lead to new useful therapies for chronic inflammatory diseases (Anderson, 1996).
Furthermore, inflammatory stimuli and inflammation can affect NF-ĸB activation, as an intracellular signaling molecule (Popivanova et al., 2008), and NF-ĸB activation is required for most immune and inflammatory responses (Roman-Blas & Jimenez, 2006). Also, studies demonstrated that NF-ĸB had an important and essential role in chronic inflammatory situations (Roman-Blas & Jimenez, 2006) and inappropriate regulation of its activity was implicated in the pathogenesis of inflammatory diseases such as RA (Roman-Blas & Jimenez, 2006). Moreover, it is described that NF-ĸB regulates genes involved in apoptosis inhibition and cell proliferation (Roman-Blas & Jimenez, 2006). It was shown that this molecule protects the cell from apoptosis (Haddad & Abdel-Karim, 2011).
Different evidence pointed that steady activation of NF-ĸB provides a κB involved in the expression variation of numerous inflammatory molecules during synoviocytes. Importance of in vivo NF-ĸB suppression in the acceleration of inflamed synovium apoptosis was discussed. They showed that in vitro, NF-κB controlled the expression of numerous inflammatory molecules in synoviocytes. Also, they demonstrated that in vivo suppression of NF-κB enhanced apoptosis in the synovium of arthritic rats. It was also found that the severity of arthritis was inhibited significantly in the contralateral untreated joints, which indicated the beneficial systemic effects of local suppression of NF-κB (Miagkov et al., 1998; Makarov, 2000).
Therefore, given the significance of apoptosis pattern change in inflammation pathophysiology, and the important role of NF-ĸB in inflammation and apoptosis induction, the current study aimed at investigating the synovial apoptosis variation during inflammation and assessing the relationship of NF-ĸB with this apoptosis variation and the inflammation caused by CFA.
2. Methods
2.1. Laboratory animals
Adult male Wistar rats, weighing 200 to 220 g were involved in the current study. The animals were kept at 22.0±1ºC under a 12-hour light-dark cycle. With the exception of testing time, food and water were available. The current study was conducted align with the investigation guidelines of the international animal models of pain and approved by Shahid Beheshti University of Medical Sciences animal studies local ethic committee (Zimmerman, 1983). The experimental groups were arranged as: CFA; CFA control; CFA+PBS; and CFA+Inh NF-ĸB. Each group was divided into 4 subgroups to assess the variations during the different days of the study (0, 7, 14, and 21) and each subgroup included 6 male rats.
2.2. Local arthritis induction
Arthritis inflammation was induced on the day 0 by a single intra-articular of 100µL CFA (10 mg/mL, CFA, Sigma, St. Louis, MO, USA) injection into the right knee joint of rats. This animal model was selected because it exhibits a rapid primary inflammation response to the adjuvant (Santora, Rasa, Visco, Steinetz, & Bagnell, 2007).
2.3. Assessment of CFA-induced arthritis
CIA development was assessed by measuring the knee diameter during different times of the study (on the days 0, 7, 14, and 21). The changes made in the knee diameter on the different days of the study were assessed using a caliper (Multag, 2012).
2.4. Synovial tissue extraction
For molecular studies, the knee joint synovial tissue of animals was removed. For this purpose, right knee joint synovia of the animals was opened and synovial membrane was removed carefully from the surrounding enclosures (Hyc, Osiecka-Iwan, Dziunycz, & Moskalewski, 2007). Then, the tissue weight was measured and recorded. At first, the obtained sample was placed in liquid nitrogen for 30 minutes. Then, it was transferred to -80ºC freezer for later use.
2.5. Measurement of synovial NF-ĸB and apoptotic markers by Western blotting
Western blot method was employed to measure the synovial level of NF-ĸB and apoptotic markers (Bax, Bcl2, and Caspase3). The synovial tissue was quickly removed after scarifying the rat and it was homogenized in lysis radioimmunoprecipitation assay (RIPA) buffer. Then, it was centrifuged at 13000 rpm (4ºC) for 25 minutes. The supernatant was separated for further analysis and the protein concentration was determined (Bradford, 1976). Equal amounts of proteins were diluted with loading (sample) buffer.
After boiling for 2 minutes, an aliquot of the diluted sample (24 µL) was loaded on electrophoresis gels. Proteins were transferred to polyvinylidene difluoride PVDF membrane (Millipore, Bedford, MA). Non-specific binding sites on the membrane were blocked by incubation in blocking buffer (2% aurora blocking agent) followed by incubation with primary antibody in blocking buffer (rabbit polyclonal IGg for Bax (1/1000), cell signaling; rabbit polyclonal IgG for Bcl2 (1/1000), cell signaling; rabbit polyclonal IgG for Caspase3 (1/1000), cell signaling; and rabbit polyclonal IgG for NF-ĸB (1/3000), abcam).
Membrane was washed 3 times with TBST (Tris-buffered saline, 0.1% Tween 20), and then, incubated (75 minutes at room temperature) with secondary antibody in blocking buffer (anti-rabbit IgG (1/3000), cell signaling for apoptotic markers; anti-rabbit IgG (1/3000), cell signaling for NF-ĸB). Membrane was, then, washed 3 times with TBST buffer. The immune reactivity of the proteins on the membrane was visualized using the chemiluminescence detection system (ECL, Amersham). The membrane was, then, incubated in stripping buffer at 37°C and incubated with β-actin primary antibody (rabbit polyclonal IgG for β-actin (1/1000), cell signaling) as a loading control. Band density was measured densitometrically using NIH Image (1.60) and expressed as the ratio of NF-ĸB, Bax, Bcl2, and Caspase3 bands to β-actin to account for any differences in starting NF-ĸB, Bax, Bcl2 and Caspase3 proteins. Each experiment was replicated 3 times with new groups of rats.
2.6. Experimental procedures
Rats were randomly divided into different experimental groups each of 24 as: CFA, CFA control, CFA+PBS and CFA+Inh-NF-ĸB. Each group was divided into 4 subgroups to assess the variations during the different days of the study (0, 7, 14, and 21) and each subgroup included 6 male rats. CIA was induced by intra-articular injection of CFA in the right knee joint by light anesthesia on the first day of study. According to the previous studies described for CFA-induced arthritis (Cicala, Innaro, & Fiorucci, 2000; Taniguchi et al., 2004), assessment of knee diameter, NF-ĸB, and the level of synovial apoptotic markers were conducted on the day 0 (immediately before CFA injection) and on the days 7 (inflammatory phase), 14, and 21 (arthritis phase).
Animals were sacrificed according to the guidelines of animal study ethics at the end of each experiment and synovial membranes were dissected, snap-frozen immediately in liquid nitrogen and kept at -80ºC. Synovial NF-ĸB and apoptotic markers level were assessed by Western blotting on the days 0, 7, 14 and 21 in the experimental groups. This procedure was also applied in the control groups.
2.7. Statistical analysis
The results of the statistical analysis were presented as mean±standard error of mean (SEM). To analyze intergroup variations, repeated measurements and one-way analysis of variance (ANOVA) followed by post hoc Tukeys multiple comparison tests in SPSS version 16 were applied. Unpaired student t test was used to determine significant differences in synovial levels of NF-ĸB and apoptotic markers between the groups. Statistical significance was considered at P≤0.05.
3. Results
3.1. Knee diameter variation during different stages of the study
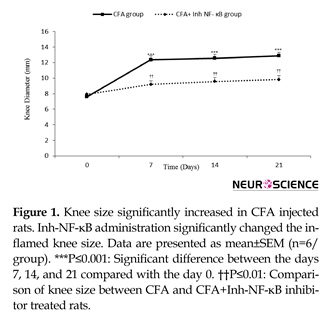
Intra-articular CFA injection increased ipsilateral knee diameter, which was continuous until the day 21 of study. Knee diameter meaningfully increased on the days 7, 14, and 21 after inflammation induction when compared with the day 0 (P≤0.001 days). The current study findings in CFA+inh-NF-ĸB group showed that inh-NF-ĸB administration caused significant reduction in knee diameter on the days 7, 14, and 21 of the study compared with those of the same days in the CFA control group (P≤0.01) (Figure 1).
There was no significant difference between the CFA injected and CFA+PBS groups in knee diameter variation during different phases of the study (results not shown). Also, there was no significant difference between the CFA injected and CFA control groups in knee diameter variation during different phases of the study (results not shown).
3.2. Synovial Bax/ Bcl2 variation during different stages of the study
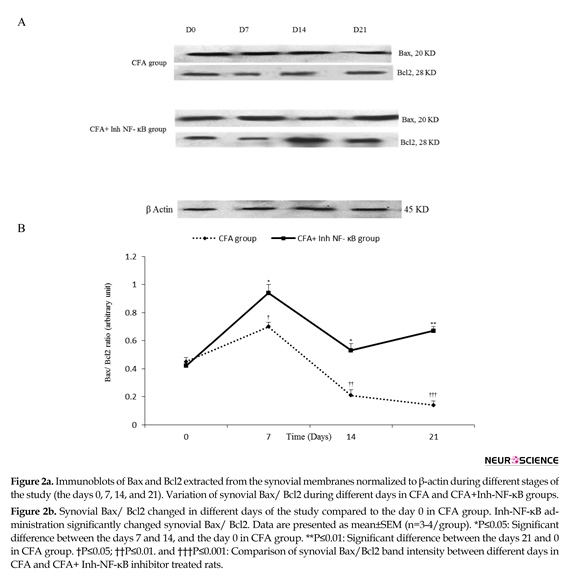
Densitometric analysis demonstrated that intra-articular CFA injection time-dependently caused significant changes in synovial Bax/ Bcl2 when compared with the control group. The current study results revealed that in synovial membranes obtained from the CFA-injected rats, synovial Bax/ Bcl2 significantly increased on the day 7 compared with the day 0 of the study (P≤0.05). On the days 14 and 21 of CFA treatment, synovial Bax/ Bcl2 were significantly lower than that of the day 0 (P≤0.05 for the day 14 and P≤0.01 for the day 21). Analysis by densitometry showed that i.p. injection of inh-NF-ĸB caused a significant increase in synovial Bax/ Bcl2 on the days 7, 14, and 21 of the study compared with those of the same days in CFA control group (P≤0.05 for the day 7, P≤0.01 for the day 14, and P≤0.001 for the day 21) (Figure 2a, b).
There was no significant difference between the CFA injected and CFA+PBS groups in synovial Bax/ Bcl2 during different phases of the study (results not shown).Based on the current study data, there was no significant difference between the CFA injected and CFA control groups in synovial Bax/ Bcl2 during different phases of the study (results not shown).
3.3. Synovial Caspase3/ β-actin variation during different stages of the study
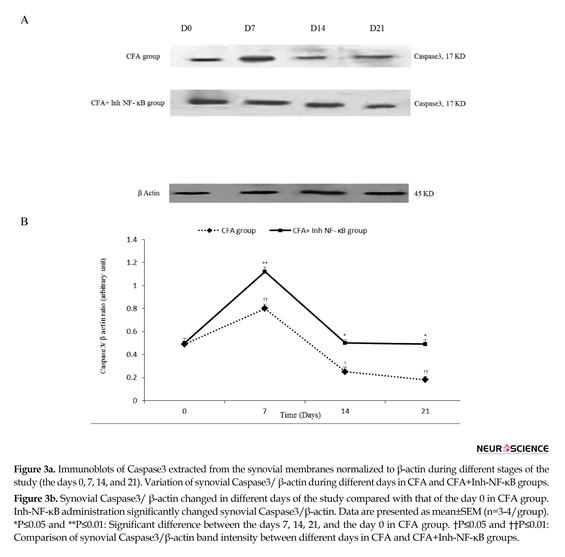
The current study results revealed that in the synovial membranes obtained from the CFA-injected rats, caspase3/ β-actin significantly raised on the day 7 (P≤0.01) and decreased on the days 14 and 21 compared to the day 0 (P≤0.05 for the day 14, P≤0.01 for the day 21). Western blot analysis showed that inh-NF-ĸB injection caused a significant increase in synovial Caspase3/ β-actin on the days 7, 14, and 21 of the study compared with those of the same days in CFA control group (P≤0.05 for the day 14, P≤0.01 for the days 7 and 21) (Figure 3a, b).
There was no significant difference between the CFA injected and CFA+PBS groups in synovial Caspase3/ β-actin during different phases of the study (results not shown). Also, there was no significant difference between the CFA injected and CFA control groups in synovial Caspase3/β-actin during different phases of the study (results not shown).
3.4. Synovial NF-ĸB/β-actin variation during different stages of the study
Western blot analysis demonstrated that intra-articular CFA injection caused significant changes in synovial NF-ĸB/β-actin when compared with those of the control
Inflammation is the primary and, essentially, important physiological reaction that normally resolves with the restoration of normal structure and function of tissue. It has symptoms such as edema, warmth, and redness (Akhtari, Zaringhalam, Eidi, Manaheji, & Tekieh, 2012; Foyet et al., 2014). Chronic inflammation is a long-duration inflammation, which can cause tissue damage and loss of function (Akhtari, et al,. 2012; Multag, 2012). Chronic inflammation is also along with increased local and systemic levels of certain inflammatory mediators such as cytokines and chemokines (Zhang, Yin, Xu, Wu, & Chen, 2012; Zaringhalam, Tekieh, Manaheji, & Akhtari, 2013). Rheumatoid Arthritis (RA) is a systemic, chronic inflammatory disorder that affects approximately 1% of the universal population. RA is described by symptoms such as joint swelling, pain, and inflammatory synovitis, which leads to hyperplasia (Kannan, Ortmann, & Kimpel, 2005; Yoshida, Ochiai, Matsuno, Panayi, & Corrigall, 2011; Foyet et al., 2014). Complete Freund’s Adjuvant (CFA)-induced arthritis is an inflammatory model commonly recommended for acute and chronic animal inflammation model (Rezazadeh, Zaringhalam, Manaheji, & Kebryaeezadeh, 2009; Zaringhalam, et al., 2013). This model is valuable to evaluate various behavioral and molecular changes made during acute and chronic inflammation due to its similarity to human RA (Taniguchi, Kanai, Kawamoto, Endo, & Higashino, 2004; Zaringhalam, et al., 2013).
Previous studies demonstrated that apoptosis dysregulation is the main pathophysiological mechanism by which mild prime inflammatory diseases progress to sever chronic inflammatory disorders (Anderson, 1996; Hayashi, 2003). Some studies demonstrated that apoptosis or programmed cell death is a central check-point, which limits the intensity of immune response (Anderson, 1996). Immune system activity and homeostasis are strongly related to apoptosis and chronic inflammation, which can be associated with apoptosis failure (Vandivier, Henson, & Douglas, 2006; Favaloro, Allocati, Graziano, Di Ilio, & De Laurenzi, 2012).
In this regard, studies showed that apoptosis impairment had an important and determinant role in the pathogenesis of lung chronic inflammatory diseases such as Acute Respiratory Distress Syndrome (ARDS), community-acquired pneumonia, Chronic Obstructive Pulmonary Disease (COPD), and Cystic Fibrosis (CF); therefore, in ARDS and community-acquired pneumonia, the number of observed apoptotic cells remain quite low, and in patients with COPD and CF apoptotic cells are increased (Vandivier et al., 2006). Moreover, studies pointed that apoptosis had an important and pivotal role in RA pathogenesis. In this regard, it was revealed that during RA, the synovial fibroblasts were resistant to apoptosis and their resistance correlated with the disease progression. Several anti-apoptotic molecules such as Sentrin‐1/small Ubiquitin‐like Modifier (SUMO)‐1, downstream modulators of Fas signaling and transcriptional regulators such as NF-κB, Stat3, and p53 are nominated to modulate apoptosis in this situation (Baier, Meineckel, Gay, & Pap, 2003; Vandivier et al, 2006; Favaloro et al., 2012; Foyet et al., 2014). Emerging body of proofs suggests that the reconnaissance of better approaches to induce selective apoptosis may lead to new useful therapies for chronic inflammatory diseases (Anderson, 1996).
Furthermore, inflammatory stimuli and inflammation can affect NF-ĸB activation, as an intracellular signaling molecule (Popivanova et al., 2008), and NF-ĸB activation is required for most immune and inflammatory responses (Roman-Blas & Jimenez, 2006). Also, studies demonstrated that NF-ĸB had an important and essential role in chronic inflammatory situations (Roman-Blas & Jimenez, 2006) and inappropriate regulation of its activity was implicated in the pathogenesis of inflammatory diseases such as RA (Roman-Blas & Jimenez, 2006). Moreover, it is described that NF-ĸB regulates genes involved in apoptosis inhibition and cell proliferation (Roman-Blas & Jimenez, 2006). It was shown that this molecule protects the cell from apoptosis (Haddad & Abdel-Karim, 2011).
Different evidence pointed that steady activation of NF-ĸB provides a κB involved in the expression variation of numerous inflammatory molecules during synoviocytes. Importance of in vivo NF-ĸB suppression in the acceleration of inflamed synovium apoptosis was discussed. They showed that in vitro, NF-κB controlled the expression of numerous inflammatory molecules in synoviocytes. Also, they demonstrated that in vivo suppression of NF-κB enhanced apoptosis in the synovium of arthritic rats. It was also found that the severity of arthritis was inhibited significantly in the contralateral untreated joints, which indicated the beneficial systemic effects of local suppression of NF-κB (Miagkov et al., 1998; Makarov, 2000).
Therefore, given the significance of apoptosis pattern change in inflammation pathophysiology, and the important role of NF-ĸB in inflammation and apoptosis induction, the current study aimed at investigating the synovial apoptosis variation during inflammation and assessing the relationship of NF-ĸB with this apoptosis variation and the inflammation caused by CFA.
2. Methods
2.1. Laboratory animals
Adult male Wistar rats, weighing 200 to 220 g were involved in the current study. The animals were kept at 22.0±1ºC under a 12-hour light-dark cycle. With the exception of testing time, food and water were available. The current study was conducted align with the investigation guidelines of the international animal models of pain and approved by Shahid Beheshti University of Medical Sciences animal studies local ethic committee (Zimmerman, 1983). The experimental groups were arranged as: CFA; CFA control; CFA+PBS; and CFA+Inh NF-ĸB. Each group was divided into 4 subgroups to assess the variations during the different days of the study (0, 7, 14, and 21) and each subgroup included 6 male rats.
2.2. Local arthritis induction
Arthritis inflammation was induced on the day 0 by a single intra-articular of 100µL CFA (10 mg/mL, CFA, Sigma, St. Louis, MO, USA) injection into the right knee joint of rats. This animal model was selected because it exhibits a rapid primary inflammation response to the adjuvant (Santora, Rasa, Visco, Steinetz, & Bagnell, 2007).
2.3. Assessment of CFA-induced arthritis
CIA development was assessed by measuring the knee diameter during different times of the study (on the days 0, 7, 14, and 21). The changes made in the knee diameter on the different days of the study were assessed using a caliper (Multag, 2012).
2.4. Synovial tissue extraction
For molecular studies, the knee joint synovial tissue of animals was removed. For this purpose, right knee joint synovia of the animals was opened and synovial membrane was removed carefully from the surrounding enclosures (Hyc, Osiecka-Iwan, Dziunycz, & Moskalewski, 2007). Then, the tissue weight was measured and recorded. At first, the obtained sample was placed in liquid nitrogen for 30 minutes. Then, it was transferred to -80ºC freezer for later use.
2.5. Measurement of synovial NF-ĸB and apoptotic markers by Western blotting
Western blot method was employed to measure the synovial level of NF-ĸB and apoptotic markers (Bax, Bcl2, and Caspase3). The synovial tissue was quickly removed after scarifying the rat and it was homogenized in lysis radioimmunoprecipitation assay (RIPA) buffer. Then, it was centrifuged at 13000 rpm (4ºC) for 25 minutes. The supernatant was separated for further analysis and the protein concentration was determined (Bradford, 1976). Equal amounts of proteins were diluted with loading (sample) buffer.
After boiling for 2 minutes, an aliquot of the diluted sample (24 µL) was loaded on electrophoresis gels. Proteins were transferred to polyvinylidene difluoride PVDF membrane (Millipore, Bedford, MA). Non-specific binding sites on the membrane were blocked by incubation in blocking buffer (2% aurora blocking agent) followed by incubation with primary antibody in blocking buffer (rabbit polyclonal IGg for Bax (1/1000), cell signaling; rabbit polyclonal IgG for Bcl2 (1/1000), cell signaling; rabbit polyclonal IgG for Caspase3 (1/1000), cell signaling; and rabbit polyclonal IgG for NF-ĸB (1/3000), abcam).
Membrane was washed 3 times with TBST (Tris-buffered saline, 0.1% Tween 20), and then, incubated (75 minutes at room temperature) with secondary antibody in blocking buffer (anti-rabbit IgG (1/3000), cell signaling for apoptotic markers; anti-rabbit IgG (1/3000), cell signaling for NF-ĸB). Membrane was, then, washed 3 times with TBST buffer. The immune reactivity of the proteins on the membrane was visualized using the chemiluminescence detection system (ECL, Amersham). The membrane was, then, incubated in stripping buffer at 37°C and incubated with β-actin primary antibody (rabbit polyclonal IgG for β-actin (1/1000), cell signaling) as a loading control. Band density was measured densitometrically using NIH Image (1.60) and expressed as the ratio of NF-ĸB, Bax, Bcl2, and Caspase3 bands to β-actin to account for any differences in starting NF-ĸB, Bax, Bcl2 and Caspase3 proteins. Each experiment was replicated 3 times with new groups of rats.
2.6. Experimental procedures
Rats were randomly divided into different experimental groups each of 24 as: CFA, CFA control, CFA+PBS and CFA+Inh-NF-ĸB. Each group was divided into 4 subgroups to assess the variations during the different days of the study (0, 7, 14, and 21) and each subgroup included 6 male rats. CIA was induced by intra-articular injection of CFA in the right knee joint by light anesthesia on the first day of study. According to the previous studies described for CFA-induced arthritis (Cicala, Innaro, & Fiorucci, 2000; Taniguchi et al., 2004), assessment of knee diameter, NF-ĸB, and the level of synovial apoptotic markers were conducted on the day 0 (immediately before CFA injection) and on the days 7 (inflammatory phase), 14, and 21 (arthritis phase).
Animals were sacrificed according to the guidelines of animal study ethics at the end of each experiment and synovial membranes were dissected, snap-frozen immediately in liquid nitrogen and kept at -80ºC. Synovial NF-ĸB and apoptotic markers level were assessed by Western blotting on the days 0, 7, 14 and 21 in the experimental groups. This procedure was also applied in the control groups.
2.7. Statistical analysis
The results of the statistical analysis were presented as mean±standard error of mean (SEM). To analyze intergroup variations, repeated measurements and one-way analysis of variance (ANOVA) followed by post hoc Tukeys multiple comparison tests in SPSS version 16 were applied. Unpaired student t test was used to determine significant differences in synovial levels of NF-ĸB and apoptotic markers between the groups. Statistical significance was considered at P≤0.05.
3. Results
3.1. Knee diameter variation during different stages of the study
Intra-articular CFA injection increased ipsilateral knee diameter, which was continuous until the day 21 of study. Knee diameter meaningfully increased on the days 7, 14, and 21 after inflammation induction when compared with the day 0 (P≤0.001 days). The current study findings in CFA+inh-NF-ĸB group showed that inh-NF-ĸB administration caused significant reduction in knee diameter on the days 7, 14, and 21 of the study compared with those of the same days in the CFA control group (P≤0.01) (Figure 1).
There was no significant difference between the CFA injected and CFA+PBS groups in knee diameter variation during different phases of the study (results not shown). Also, there was no significant difference between the CFA injected and CFA control groups in knee diameter variation during different phases of the study (results not shown).
3.2. Synovial Bax/ Bcl2 variation during different stages of the study
Densitometric analysis demonstrated that intra-articular CFA injection time-dependently caused significant changes in synovial Bax/ Bcl2 when compared with the control group. The current study results revealed that in synovial membranes obtained from the CFA-injected rats, synovial Bax/ Bcl2 significantly increased on the day 7 compared with the day 0 of the study (P≤0.05). On the days 14 and 21 of CFA treatment, synovial Bax/ Bcl2 were significantly lower than that of the day 0 (P≤0.05 for the day 14 and P≤0.01 for the day 21). Analysis by densitometry showed that i.p. injection of inh-NF-ĸB caused a significant increase in synovial Bax/ Bcl2 on the days 7, 14, and 21 of the study compared with those of the same days in CFA control group (P≤0.05 for the day 7, P≤0.01 for the day 14, and P≤0.001 for the day 21) (Figure 2a, b).
There was no significant difference between the CFA injected and CFA+PBS groups in synovial Bax/ Bcl2 during different phases of the study (results not shown).Based on the current study data, there was no significant difference between the CFA injected and CFA control groups in synovial Bax/ Bcl2 during different phases of the study (results not shown).
3.3. Synovial Caspase3/ β-actin variation during different stages of the study
The current study results revealed that in the synovial membranes obtained from the CFA-injected rats, caspase3/ β-actin significantly raised on the day 7 (P≤0.01) and decreased on the days 14 and 21 compared to the day 0 (P≤0.05 for the day 14, P≤0.01 for the day 21). Western blot analysis showed that inh-NF-ĸB injection caused a significant increase in synovial Caspase3/ β-actin on the days 7, 14, and 21 of the study compared with those of the same days in CFA control group (P≤0.05 for the day 14, P≤0.01 for the days 7 and 21) (Figure 3a, b).
There was no significant difference between the CFA injected and CFA+PBS groups in synovial Caspase3/ β-actin during different phases of the study (results not shown). Also, there was no significant difference between the CFA injected and CFA control groups in synovial Caspase3/β-actin during different phases of the study (results not shown).
3.4. Synovial NF-ĸB/β-actin variation during different stages of the study
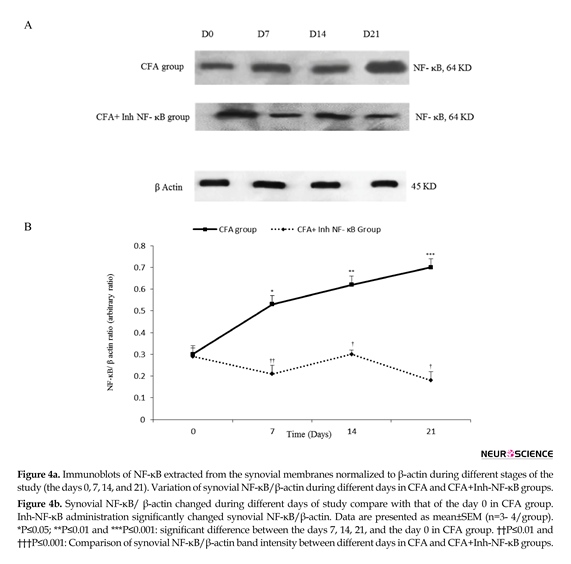
Western blot analysis demonstrated that intra-articular CFA injection caused significant changes in synovial NF-ĸB/β-actin when compared with those of the control


group. The current study findings showed that among the synovial membranes acquired from the CFA injected rats, synovial NF-ĸB/β-actin significantly increased on the days 7, 14, and 21 when compared with those of the day 0 of the study (P≤0.05 for the day 7, P≤0.01 for the day 14, and P≤0.001 for the day 21). Densitometric analysis showed that inh-NF-ĸB injection caused a significant reduction in synovial NF-ĸB/β-actin on the days 7, 14, and 21 of the study compared with those of the same days in CFA control group (P≤0.01 for the days 7 and 14, P≤0.001 for the day 21) (Figure 4a, b).
There was no significant difference between the CFA injected and CFA+PBS groups in synovial NF-ĸB/β-actin during different phases of the study (results not shown). Also, there was no significant difference between the CFA injected and CFA control groups in synovial NF-ĸB/β-actin during different phases of the study (results not shown).
4. Discussion
The current study results demonstrated that intra-articular CFA injection caused permanent synovial edema. Findings also pointed out that there was a relationship between synovial apoptosis pattern change during the study period and inflammation progression. Also, it was shown that synovial NF-ĸB had an important role in synovial apoptosis change, which can describe a part of the relationship between CFA-inflammation progress and apoptosis.
CFA injection in rat knee joint caused permanent synovial edema during the whole study period. Studies showed
There was no significant difference between the CFA injected and CFA+PBS groups in synovial NF-ĸB/β-actin during different phases of the study (results not shown). Also, there was no significant difference between the CFA injected and CFA control groups in synovial NF-ĸB/β-actin during different phases of the study (results not shown).
4. Discussion
The current study results demonstrated that intra-articular CFA injection caused permanent synovial edema. Findings also pointed out that there was a relationship between synovial apoptosis pattern change during the study period and inflammation progression. Also, it was shown that synovial NF-ĸB had an important role in synovial apoptosis change, which can describe a part of the relationship between CFA-inflammation progress and apoptosis.
CFA injection in rat knee joint caused permanent synovial edema during the whole study period. Studies showed

that local CFA injection was one of the most scientific models of inflammatory disorders (like RA) induction and this model was useful to assess various behavioral and molecular changes made during the inflammatory arthritis (Wang & Shuaib, 2002). The authors’ previous studies also showed that plantar CFA injection induced constant inflammation during the 21 days of the study (Akhtari, Zaringhalam, Eidi, Manaheji, Manaheji, & Tekieh, 2015).
On the other hand, the current study findings revealed that CFA-induced inflammation caused an increase in synovial apoptotic markers in the acute phase of the study, but these markers decreased during the chronic phase. The current study results in acute phase of inflammation aligned with those of some previous studies. Among them, it can be pointed that alveolar epithelial cells become apoptotic in acute lung inflammation; therefore, inflammatory mediators in this situation cause apoptosis induction (Matute-Bello, et al., 2001).
Anothers’ study showed that during acute renal inflammation caused by acute ischemic or nephrotoxic insult, apoptosis increases (Lieberthal, 1997). Aligned with the current study results during the chronic phase of inflammation, studies showed that chronic inflammatory bowel diseases can induce cell resistance to apoptosis, and hence, prone the patient to cancer (Shacter & Weitzman, 2002). Previous studies showed that inflammatory disorders can affect apoptosis pattern and also, apoptosis change can affect inflammatory contexts; therefore, apoptosis failure can lead to auto-immune diseases (such as RA), and induction of apoptotic pathways may play
On the other hand, the current study findings revealed that CFA-induced inflammation caused an increase in synovial apoptotic markers in the acute phase of the study, but these markers decreased during the chronic phase. The current study results in acute phase of inflammation aligned with those of some previous studies. Among them, it can be pointed that alveolar epithelial cells become apoptotic in acute lung inflammation; therefore, inflammatory mediators in this situation cause apoptosis induction (Matute-Bello, et al., 2001).
Anothers’ study showed that during acute renal inflammation caused by acute ischemic or nephrotoxic insult, apoptosis increases (Lieberthal, 1997). Aligned with the current study results during the chronic phase of inflammation, studies showed that chronic inflammatory bowel diseases can induce cell resistance to apoptosis, and hence, prone the patient to cancer (Shacter & Weitzman, 2002). Previous studies showed that inflammatory disorders can affect apoptosis pattern and also, apoptosis change can affect inflammatory contexts; therefore, apoptosis failure can lead to auto-immune diseases (such as RA), and induction of apoptotic pathways may play

important roles in the improvement of inflammatory symptoms (Ramos et al., 2010; Zhang, et al,. 2012).
To the authors’ best knowledge, it was confirmed that inhibition of increased apoptosis in some chronic inflammatory disorders can result in fortification of inflammatory symptoms (Papadakis & Targan, 2000). Moreover, the current study results showed that articular CFA injection caused an increase in synovial NF-ĸB level during the 21-day study period and Inh-NF-ĸB decreased knee diameter in the whole study period. It was shown that incorrectly regulation of NF- ĸB associated with conditions such as autoimmune and inflammatory diseases (Freudenthal, et al., 1998; Merlo, Freudenthal, & Romano, 2002; Meffert, Chang, Wiltgen, Fanselow, & Baltimore, 2003).
Previous studies indicated that NF-ĸB is one of the principal intra-cellular signaling molecules that mediate inflammatory mediators’ effects (Libermann & Baltimore, 1990; Garcia et al., 2000), and it was also shown that inflammatory situations affect NF-ĸB activation (Popivanova et al., 2008). Considering the important role of NF- ĸB in advancing processes that lead to the development of inflammation, it is predictable that inhibition of NF-ĸB can improve inflammation and its related symptoms such as edema.
To the authors’ best knowledge, it was confirmed that inhibition of increased apoptosis in some chronic inflammatory disorders can result in fortification of inflammatory symptoms (Papadakis & Targan, 2000). Moreover, the current study results showed that articular CFA injection caused an increase in synovial NF-ĸB level during the 21-day study period and Inh-NF-ĸB decreased knee diameter in the whole study period. It was shown that incorrectly regulation of NF- ĸB associated with conditions such as autoimmune and inflammatory diseases (Freudenthal, et al., 1998; Merlo, Freudenthal, & Romano, 2002; Meffert, Chang, Wiltgen, Fanselow, & Baltimore, 2003).
Previous studies indicated that NF-ĸB is one of the principal intra-cellular signaling molecules that mediate inflammatory mediators’ effects (Libermann & Baltimore, 1990; Garcia et al., 2000), and it was also shown that inflammatory situations affect NF-ĸB activation (Popivanova et al., 2008). Considering the important role of NF- ĸB in advancing processes that lead to the development of inflammation, it is predictable that inhibition of NF-ĸB can improve inflammation and its related symptoms such as edema.
Furthermore, the current study findings demonstrated that when CFA induced inflammation, increase in synovial NF-ĸB level was contrary to the variance of synovial apoptotic markers. Also, the current study showed that inhibition of NF-ĸB activity caused an elevation in synovial apoptotic markers during the whole study period. These results indicated that NF-ĸB had an anti-apoptotic effect on synovial tissue during CFA-induced inflammation. Consistent with the current study results, previous studies pointed out the anti-apoptotic effects of NF-ĸB. It was shown that in human chondrocytes, NF-ĸB activation mediated anti-apoptotic effects (Roman-Blas & Jimenez, 2006). Also, studies illustrated that defect in NF-ĸB activity caused increased susceptibility to apoptosis (Haddad & Abdel-Karim, 2011). Accordingly, it seems that the decrease in apoptosis in chronic inflammation is associated with increased activity of NF-ĸB.
Therefore, according to the results of the current study, a relationship between CFA-induced inflammation and apoptosis variation can be suggested, and it seems that the decrease of apoptotic markers during chronic inflammation can be considered as one of the important reasons of inflammation continuation. On the other hand, the current study results revealed the crucial role of synovial NF-ĸB in inhibition of apoptosis during chronic inflammation and it seems that treatment by Inh-NF-ĸB can be suggested as an important method to decrease symptoms of persistent inflammation, which needs more investigations.
Acknowledgments
The current article was based on data obtained from Sahar Golabi PhD thesis, which was done in Neurophysiology Research Center of Shahid Beheshti University of Medical Sciences, Tehran, Iran.
Conflict of Interest
The authors declared no conflicts of interest.
References
Akhtari, Z., Zaringhalam, J., Eidi, A., Manaheji, H., & Tekieh, E. (2012). Bidirectional effects of serum TNF alpha level and spinal p38MAPK phosphorylation on hyperalgesia variation during CFA-induced arthritis. EXCLI Journal, 11, 373-85. PMCID: PMC4941798
Akhtari, Z., Zaringhalam, J., Eidi, A., Manaheji, H., Manaheji, H., & Tekieh, E. (2015). [Relation cellular- molecular between serum IL10 levels and hyperalgesia variation in adjuvant- induced arthritis (Persian)]. Iranian South Medical Journal; 17(6), 1176-1187.
Anderson, G. P. (1996). Resolution of chronic inflammation by therapeutic induction of apoptosis. Trends in Pharmacological Sciences, 17(12), 438–442. doi: 10.1016/s0165-6147(96)01004-8
Baier, A., Meineckel, I., Gay, S., & Pap, T. (2003). Apoptosis in rheumatoid arthritis. Current Opinion in Rheumatology, 15(3), 274–9. doi: 10.1097/00002281-200305000-00015
Bradford, M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72(1-2), 248–54. doi: 10.1006/abio.1976.9999
Cicala, C., Innaro, A. & Fiorucci, S. (2000). No-naproxen modulates inflammation, nociception and downregulates T cell response in rat Freunds Adjuvant Arthiritis. British Journal of Pharmacology, 130(6), 490-7. doi: 10.1038/sj.bjp.0703449
Favaloro, B., Allocati, N., Graziano, V., Di Ilio, C., & De Laurenzi, V. (2012). Role of Apoptosis in disease. Aging, 4(5), 330–49. doi: 10.18632/aging.100459
Foyet, H. S., Tsala, D. E., Bodo, J. C. Z. E., Carine, A. N., Fabrice, N. H., Heroyne, L. T., et al. (2014). Anti-arthritic and anti-inflammatory propensity of catechins from Vitellaria paradoxa. Pharmacognosy Research, 7(4), 367-77. PMCID: PMC4660517
Freudenthal, R., Locatelli, F., Hermitte, G., Maldonado, H., Lafourcade, C., Delorenzi, A., et al. (1998). κ-B like DNA-binding activity is enhanced after spaced training that induces long-term memory in the crab Chasmagnathus. Neuroscience Letters, 242(3), 143–6. doi: 10.1016/s0304-3940(98)00059-7
Garcia, G. E., Xia, Y., Chen, S., Wang, Y., Ye, R. D., Harrison, J. K., et al. (2000). NF-kappa B-dependent Fractalkine induction in rat aortic endothelial cells stimulated by IL-1beta, TNF-alpha, and LPS. Journal of Leukocyte Biology, 67(4), 577-84. PMID: 10770292
Haddad, J. J., & Abdel-Karim, N. E. (2011). NF-κB cellular and molecular regulatory mechanisms and pathways: Therapeutic pattern or pseudoregulation? Cellular Immunology, 271(1), 5–14. doi: 10.1016/j.cellimm.2011.06.021
Hayashi, T. (2003). Role of defective apoptosis in type 1 diabetes and other autoimmune diseases. Recent Progress in Hormone Research, 58(1), 131–53. doi: 10.1210/rp.58.1.131
Hyc, A., Osiecka-Iwan, A., Dziunycz, P., & Moskalewski, S. (2007). Preparation of rat synovial membrane for studies of cytokine secretion. Folia Histochemica and Cytobiologica, 45(1), 57-60. PMID: 17378248
Kannan, K., Ortmann, R. A., & Kimpel, D. (2005). Animal models of rheumatoid arthritis and their relevance to human disease. Pathophysiology, 12(3), 167–81. doi: 10.1016/j.pathophys.2005.07.011
Libermann, T. A., & Baltimore, D. (1990). Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Molecular and Cellular Biology, 10(5), 2327–34. doi: 10.1128/mcb.10.5.2327
Lieberthal, W. (1997). Biology of acute renal failure: Therapeutic implications. Kidney International, 52(4), 1102–15. doi: 10.1038/ki.1997.435
Makarov, S. S. (2000). NF-κB as a therapeutic target in chronic inflammation: recent advances. Molecular Medicine Today, 6(11), 441–8. doi: 10.1016/s1357-4310(00)01814-1
Matute-Bello, G., Winn, R. K., Jonas, M., Chi, E. Y., Martin, T. R., & Liles, W. C. (2001). Fas (CD95)induces alveolar epithelial cell apoptosis in vivo. The American Journal of Pathology, 158(1), 153–61. doi: 10.1016/s0002-9440(10)63953-3
Therefore, according to the results of the current study, a relationship between CFA-induced inflammation and apoptosis variation can be suggested, and it seems that the decrease of apoptotic markers during chronic inflammation can be considered as one of the important reasons of inflammation continuation. On the other hand, the current study results revealed the crucial role of synovial NF-ĸB in inhibition of apoptosis during chronic inflammation and it seems that treatment by Inh-NF-ĸB can be suggested as an important method to decrease symptoms of persistent inflammation, which needs more investigations.
Acknowledgments
The current article was based on data obtained from Sahar Golabi PhD thesis, which was done in Neurophysiology Research Center of Shahid Beheshti University of Medical Sciences, Tehran, Iran.
Conflict of Interest
The authors declared no conflicts of interest.
References
Akhtari, Z., Zaringhalam, J., Eidi, A., Manaheji, H., & Tekieh, E. (2012). Bidirectional effects of serum TNF alpha level and spinal p38MAPK phosphorylation on hyperalgesia variation during CFA-induced arthritis. EXCLI Journal, 11, 373-85. PMCID: PMC4941798
Akhtari, Z., Zaringhalam, J., Eidi, A., Manaheji, H., Manaheji, H., & Tekieh, E. (2015). [Relation cellular- molecular between serum IL10 levels and hyperalgesia variation in adjuvant- induced arthritis (Persian)]. Iranian South Medical Journal; 17(6), 1176-1187.
Anderson, G. P. (1996). Resolution of chronic inflammation by therapeutic induction of apoptosis. Trends in Pharmacological Sciences, 17(12), 438–442. doi: 10.1016/s0165-6147(96)01004-8
Baier, A., Meineckel, I., Gay, S., & Pap, T. (2003). Apoptosis in rheumatoid arthritis. Current Opinion in Rheumatology, 15(3), 274–9. doi: 10.1097/00002281-200305000-00015
Bradford, M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72(1-2), 248–54. doi: 10.1006/abio.1976.9999
Cicala, C., Innaro, A. & Fiorucci, S. (2000). No-naproxen modulates inflammation, nociception and downregulates T cell response in rat Freunds Adjuvant Arthiritis. British Journal of Pharmacology, 130(6), 490-7. doi: 10.1038/sj.bjp.0703449
Favaloro, B., Allocati, N., Graziano, V., Di Ilio, C., & De Laurenzi, V. (2012). Role of Apoptosis in disease. Aging, 4(5), 330–49. doi: 10.18632/aging.100459
Foyet, H. S., Tsala, D. E., Bodo, J. C. Z. E., Carine, A. N., Fabrice, N. H., Heroyne, L. T., et al. (2014). Anti-arthritic and anti-inflammatory propensity of catechins from Vitellaria paradoxa. Pharmacognosy Research, 7(4), 367-77. PMCID: PMC4660517
Freudenthal, R., Locatelli, F., Hermitte, G., Maldonado, H., Lafourcade, C., Delorenzi, A., et al. (1998). κ-B like DNA-binding activity is enhanced after spaced training that induces long-term memory in the crab Chasmagnathus. Neuroscience Letters, 242(3), 143–6. doi: 10.1016/s0304-3940(98)00059-7
Garcia, G. E., Xia, Y., Chen, S., Wang, Y., Ye, R. D., Harrison, J. K., et al. (2000). NF-kappa B-dependent Fractalkine induction in rat aortic endothelial cells stimulated by IL-1beta, TNF-alpha, and LPS. Journal of Leukocyte Biology, 67(4), 577-84. PMID: 10770292
Haddad, J. J., & Abdel-Karim, N. E. (2011). NF-κB cellular and molecular regulatory mechanisms and pathways: Therapeutic pattern or pseudoregulation? Cellular Immunology, 271(1), 5–14. doi: 10.1016/j.cellimm.2011.06.021
Hayashi, T. (2003). Role of defective apoptosis in type 1 diabetes and other autoimmune diseases. Recent Progress in Hormone Research, 58(1), 131–53. doi: 10.1210/rp.58.1.131
Hyc, A., Osiecka-Iwan, A., Dziunycz, P., & Moskalewski, S. (2007). Preparation of rat synovial membrane for studies of cytokine secretion. Folia Histochemica and Cytobiologica, 45(1), 57-60. PMID: 17378248
Kannan, K., Ortmann, R. A., & Kimpel, D. (2005). Animal models of rheumatoid arthritis and their relevance to human disease. Pathophysiology, 12(3), 167–81. doi: 10.1016/j.pathophys.2005.07.011
Libermann, T. A., & Baltimore, D. (1990). Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Molecular and Cellular Biology, 10(5), 2327–34. doi: 10.1128/mcb.10.5.2327
Lieberthal, W. (1997). Biology of acute renal failure: Therapeutic implications. Kidney International, 52(4), 1102–15. doi: 10.1038/ki.1997.435
Makarov, S. S. (2000). NF-κB as a therapeutic target in chronic inflammation: recent advances. Molecular Medicine Today, 6(11), 441–8. doi: 10.1016/s1357-4310(00)01814-1
Matute-Bello, G., Winn, R. K., Jonas, M., Chi, E. Y., Martin, T. R., & Liles, W. C. (2001). Fas (CD95)induces alveolar epithelial cell apoptosis in vivo. The American Journal of Pathology, 158(1), 153–61. doi: 10.1016/s0002-9440(10)63953-3
Meffert, M. K., Chang, J. M., Wiltgen, B. J., Fanselow, M. S., & Baltimore, D. (2003). NF-κB functions in synaptic signaling and behavior. Nature Neuroscience, 6(10), 1072–8. doi: 10.1038/nn1110
Merlo, E., Freudenthal, R., & Romano, A. (2002). The IκB kinase inhibitor sulfasalazine impairs long-term memory in the crab Chasmagnathus. Neuroscience, 112(1), 161–72. doi: 10.1016/s0306-4522(02)00049-0
Miagkov, A. V., Kovalenko, D. V., Brown, C. E., Didsbury, J. R., Cogswell, J. P., Stimpson, S. A., et al. (1998). NF-B activation provides the potential link between inflammation and hyperplasia in the arthritic joint. Proceedings of the National Academy of Sciences, 95(23), 13859–64. doi: 10.1073/pnas.95.23.13859
Multag, S. H. (2012). [Dose dependent anti-inflammatory effect of Ammi majus alcoholic extract in rat: chronic study (Arabic)]. Iraqi Journal of Pharmaceutical Sciences, 21, 82-86.
Ramos, M. V., Fernández, G. C., Brando, R. J. F., Panek, C. A., Bentancor, L. V., Landoni, V. I., et al. (2009). Interleukin-10 and interferon-γ modulate surface expression of fractalkine-receptor (CX3CR1) via PI3K in monocytes. Immunology, 129(4), 600–09. doi: 10.1111/j.1365-2567.2009.03181.x
Papadakis, K. A., & Targan, S. R. (2000). Tumor necrosis factor: Biology and therapeutic inhibitors. Gastroenterology, 119(4), 1148–57. doi: 10.1053/gast.2000.18160
Popivanova, B. K., Kitamura, K., Wu, Y., Kondo, T., Kagaya, T., Kaneko, S., et al. (2008). Blocking TNF-α in mice reduces colorectal carcinogenesis associated with chronic colitis. Journal of Clinical Investigation. 118(2), 560-70. doi: 10.1172/jci32453
Rezazadeh, S. H., Zaringhalam, J., Manaheji, H., & Kebryaeezadeh, A. (2009). Anti-inflammatory and anti–hyperalgesia activities of Stachys athorecalyx extracts on CFA-induced inflammation. Journal of Medicinal Plants Research, 3(5), 368-376.
Roman-Blas, J. A., & Jimenez, S. A. (2006). NF-κB as a potential therapeutic target in osteoarthritis and rheumatoid arthritis. Osteoarthritis and Cartilage, 14(9), 839–48. doi: 10.1016/j.joca.2006.04.008
Santora, K., Rasa, C., Visco, D., Steinetz, B. G., & Bagnell, C. A. (2007). Antiarthritic effects of relaxin, in combination with estrogen, in rat adjuvant-induced arthritis. Journal of Pharmacology and Experimental Therapeutics, 322(2), 887–93. doi: 10.1124/jpet.107.122903
Shacter, E., & Weitzman, S. A. (2002). Chronic inflammation and cancer. Oncology, 16(2), 217-26. PMID: 11866137
Taniguchi, N., Kanai, S., Kawamoto, M., Endo, H., & Higashino, H. (2004). Study onapplication of static magnetic field for adjuvant arthritis rats. Evidence-Based Complementary and Alternative Medicine, 1(2), 187–91. doi: 10.1093/ecam/neh024
Wang, C. X., & Shuaib, A. (2002). Involvement of inflammatory cytokines in central nervous system injury. Progress in Neurobiology, 67(2), 161–72. doi: 10.1016/s0301-0082(02)00010-2
Vandivier, R. W., Henson, P. M., & Douglas, I. S. (2006). Burying the dead: The impact of failed apoptotic cell removal (Efferocytosis) on chronic inflammatory lung disease. Chest, 129(6), 1673–82. doi: 10.1378/chest.129.6.1673
Yoshida, K., Ochiai, A., Matsuno, H., Panayi, G. S., & Corrigall, V. M. (2011). Binding immunoglobulin protein resolves rheumatoid synovitis: a xenogeneic study using rheumatoid arthritis synovial membrane transplants in SCID mice. Arthritis Research & Therapy, 13(5), 149. doi: 10.1186/ar3463
Zaringhalam, J., Tekieh, E., Manaheji, H., & Akhtari, Z. (2013). Cellular events during arthritis-induced hyperalgesia are mediated by Interleukin-6 and p38 MAPK and their effects on the expression of spinal mu-opioid receptors. Rheumatology International, 33(9), 2291–9. doi: 10.1007/s00296-013-2715-2
Zhang, N., Yin, Y., Xu, S. H. J., Wu, Y. P., & Chen, W. S. H. (2012). Inflammation and apoptosis in spinal cord injury. Indian Journal of Medical Research, 135, 287-96. PMCID: PMC3361863
Zimmermann, M. (1983). Ethical guidelines for investigations of experimental pain in conscious animals. Pain, 16(2), 109–10. doi: 10.1016/0304-3959(83)90201-4
Merlo, E., Freudenthal, R., & Romano, A. (2002). The IκB kinase inhibitor sulfasalazine impairs long-term memory in the crab Chasmagnathus. Neuroscience, 112(1), 161–72. doi: 10.1016/s0306-4522(02)00049-0
Miagkov, A. V., Kovalenko, D. V., Brown, C. E., Didsbury, J. R., Cogswell, J. P., Stimpson, S. A., et al. (1998). NF-B activation provides the potential link between inflammation and hyperplasia in the arthritic joint. Proceedings of the National Academy of Sciences, 95(23), 13859–64. doi: 10.1073/pnas.95.23.13859
Multag, S. H. (2012). [Dose dependent anti-inflammatory effect of Ammi majus alcoholic extract in rat: chronic study (Arabic)]. Iraqi Journal of Pharmaceutical Sciences, 21, 82-86.
Ramos, M. V., Fernández, G. C., Brando, R. J. F., Panek, C. A., Bentancor, L. V., Landoni, V. I., et al. (2009). Interleukin-10 and interferon-γ modulate surface expression of fractalkine-receptor (CX3CR1) via PI3K in monocytes. Immunology, 129(4), 600–09. doi: 10.1111/j.1365-2567.2009.03181.x
Papadakis, K. A., & Targan, S. R. (2000). Tumor necrosis factor: Biology and therapeutic inhibitors. Gastroenterology, 119(4), 1148–57. doi: 10.1053/gast.2000.18160
Popivanova, B. K., Kitamura, K., Wu, Y., Kondo, T., Kagaya, T., Kaneko, S., et al. (2008). Blocking TNF-α in mice reduces colorectal carcinogenesis associated with chronic colitis. Journal of Clinical Investigation. 118(2), 560-70. doi: 10.1172/jci32453
Rezazadeh, S. H., Zaringhalam, J., Manaheji, H., & Kebryaeezadeh, A. (2009). Anti-inflammatory and anti–hyperalgesia activities of Stachys athorecalyx extracts on CFA-induced inflammation. Journal of Medicinal Plants Research, 3(5), 368-376.
Roman-Blas, J. A., & Jimenez, S. A. (2006). NF-κB as a potential therapeutic target in osteoarthritis and rheumatoid arthritis. Osteoarthritis and Cartilage, 14(9), 839–48. doi: 10.1016/j.joca.2006.04.008
Santora, K., Rasa, C., Visco, D., Steinetz, B. G., & Bagnell, C. A. (2007). Antiarthritic effects of relaxin, in combination with estrogen, in rat adjuvant-induced arthritis. Journal of Pharmacology and Experimental Therapeutics, 322(2), 887–93. doi: 10.1124/jpet.107.122903
Shacter, E., & Weitzman, S. A. (2002). Chronic inflammation and cancer. Oncology, 16(2), 217-26. PMID: 11866137
Taniguchi, N., Kanai, S., Kawamoto, M., Endo, H., & Higashino, H. (2004). Study onapplication of static magnetic field for adjuvant arthritis rats. Evidence-Based Complementary and Alternative Medicine, 1(2), 187–91. doi: 10.1093/ecam/neh024
Wang, C. X., & Shuaib, A. (2002). Involvement of inflammatory cytokines in central nervous system injury. Progress in Neurobiology, 67(2), 161–72. doi: 10.1016/s0301-0082(02)00010-2
Vandivier, R. W., Henson, P. M., & Douglas, I. S. (2006). Burying the dead: The impact of failed apoptotic cell removal (Efferocytosis) on chronic inflammatory lung disease. Chest, 129(6), 1673–82. doi: 10.1378/chest.129.6.1673
Yoshida, K., Ochiai, A., Matsuno, H., Panayi, G. S., & Corrigall, V. M. (2011). Binding immunoglobulin protein resolves rheumatoid synovitis: a xenogeneic study using rheumatoid arthritis synovial membrane transplants in SCID mice. Arthritis Research & Therapy, 13(5), 149. doi: 10.1186/ar3463
Zaringhalam, J., Tekieh, E., Manaheji, H., & Akhtari, Z. (2013). Cellular events during arthritis-induced hyperalgesia are mediated by Interleukin-6 and p38 MAPK and their effects on the expression of spinal mu-opioid receptors. Rheumatology International, 33(9), 2291–9. doi: 10.1007/s00296-013-2715-2
Zhang, N., Yin, Y., Xu, S. H. J., Wu, Y. P., & Chen, W. S. H. (2012). Inflammation and apoptosis in spinal cord injury. Indian Journal of Medical Research, 135, 287-96. PMCID: PMC3361863
Zimmermann, M. (1983). Ethical guidelines for investigations of experimental pain in conscious animals. Pain, 16(2), 109–10. doi: 10.1016/0304-3959(83)90201-4
Type of Study: Original |
Subject:
Behavioral Neuroscience
Received: 2016/11/9 | Accepted: 2017/02/3 | Published: 2017/05/1
Received: 2016/11/9 | Accepted: 2017/02/3 | Published: 2017/05/1
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
