

Volume 16, Issue 2 (March & April 2025)
BCN 2025, 16(2): 505-518 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Bayat S, Gholami M, Khodadadi H, Ghazavi M, Nasiri J, Kheirollahi M. Expanding the Phenotype and Genotype Spectrum of a Novel Mutation in Hypomyelinating Leukodystrophy-5 With a Review of the Literature on 42 Cases. BCN 2025; 16 (2) :505-518
URL: http://bcn.iums.ac.ir/article-1-3026-en.html
URL: http://bcn.iums.ac.ir/article-1-3026-en.html
Sahar Bayat1 

 , Milad Gholami2 , Hamidreza Khodadadi3 , Mohammadreza Ghazavi4 , Jafar Nasiri5 , Majid Kheirollahi *1
, Milad Gholami2 , Hamidreza Khodadadi3 , Mohammadreza Ghazavi4 , Jafar Nasiri5 , Majid Kheirollahi *1
, Milad Gholami2 , Hamidreza Khodadadi3 , Mohammadreza Ghazavi4 , Jafar Nasiri5 , Majid Kheirollahi *1
1- Department of Genetics and Molecular Biology, Faculty of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran.
2- Department of Biochemistry and Genetics, School of Medicine, Arak University of Medical Sciences, Arāk, Iran.
3- Department of Biotechnology, School of Medicine, Lorestan University of Medical Sciences, Lorestan, Iran.
4- Department of Pediatric Neurology, Faculty of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran.
5- Department of Pediatric Neurology, Faculty of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran. & Child Growth and Development Research Center, Isfahan University of Medical Sciences, Isfahan, Iran.
2- Department of Biochemistry and Genetics, School of Medicine, Arak University of Medical Sciences, Arāk, Iran.
3- Department of Biotechnology, School of Medicine, Lorestan University of Medical Sciences, Lorestan, Iran.
4- Department of Pediatric Neurology, Faculty of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran.
5- Department of Pediatric Neurology, Faculty of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran. & Child Growth and Development Research Center, Isfahan University of Medical Sciences, Isfahan, Iran.
Keywords: Hypomyelinating leukodystrophy-5 (HLD-5), FAM126A, DRCTNNB1A, HYCC1, Whole exome sequencing (WES)
Full-Text [PDF 1126 kb]
| Abstract (HTML)
Full-Text:
1. Introduction
Myelin sheath around axons is formed by the spiral wrapping of the differentiated oligodendroglial cell plasma membrane in the central nervous system (CNS) and Schwann cell plasma membrane in the peripheral nervous system (PNS) (Simons & Nave, 2016). These myelin sheaths play an intricate role in saltatory conduction and protecting axons from cellular stresses (Nave & Werner, 2021). Hypomyelinating leukodystrophy (HLD) is a group of genetic demyelinating or dysmyelinating diseases that affect the proper development of the myelin sheath in the CNS (Charzewska et al., 2016).
HLDs are generally rare, heterogeneous, and challenging to diagnose because they have diverse clinical manifestations that most often include neurological dysfunction such as ataxia, motor ability, and intellectual disability (Charzewska et al., 2016). Next-generation sequencing (NGS) has revolutionized the potential to identify multiple autosomal recessive HLD genes (Torii & Yamauchi, 2023).
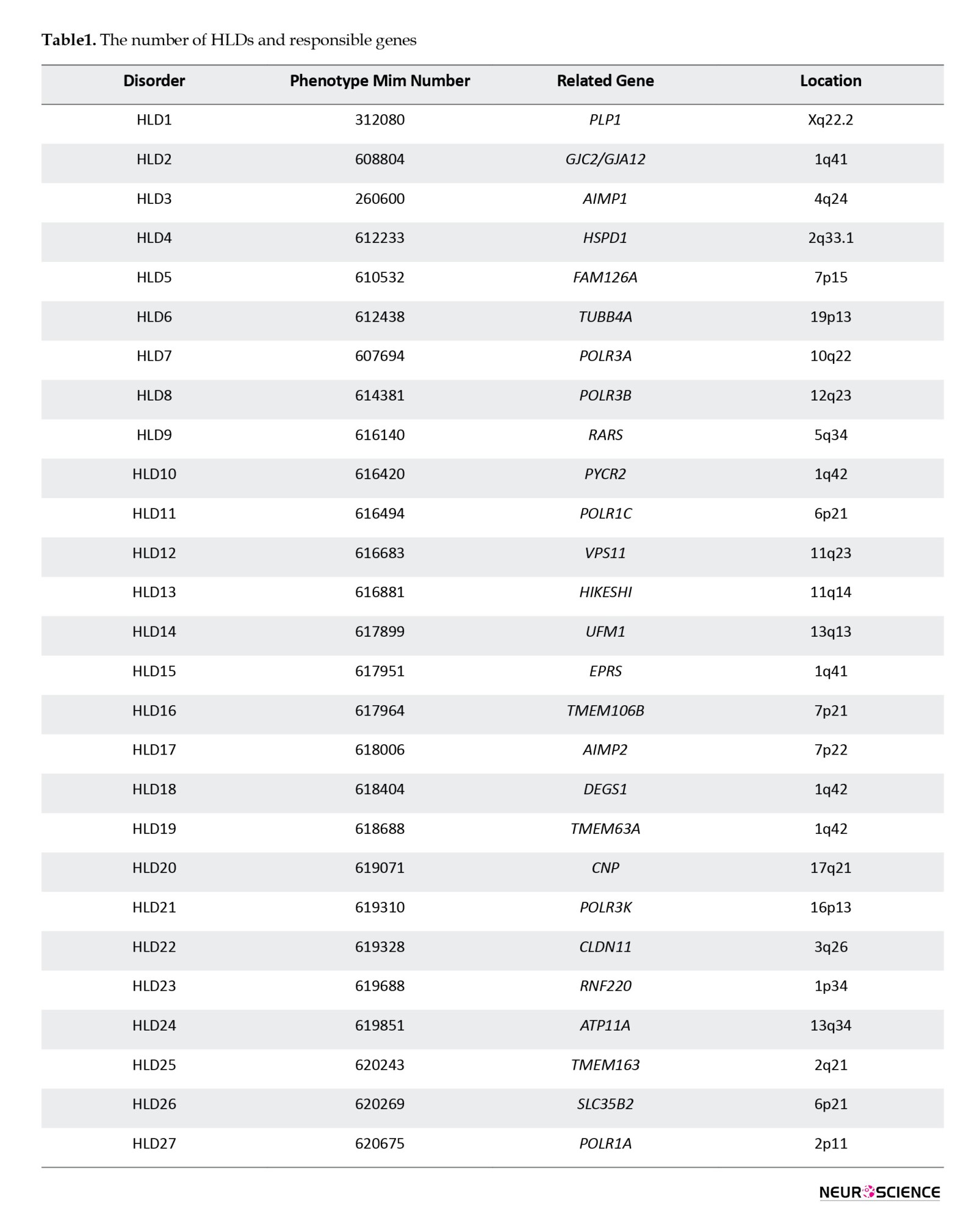
According to the Online Mendelian Inheritance in Man (OMIM, 2025), the number of HLDs and responsible genes identified in the last 10 years has increased (Table 1).
Among HLDs, HLD-5 is a rare genetic disorder caused by an autosomal recessive mutation in the FAM126A (formerly DRCTNNB1A) gene (Torii & Yamauchi, 2023). The FAM126A gene, also known as hyccin PI4KA lipid kinase complex subunit 1 (HYCC1), is located on chromosome 7p15.3, which contains 14 exons and 13 introns. This gene encodes a protein containing 521 amino acids (National Library of Medicine, 2025).
FAM126A expression is downregulated by beta-catenin, an essential protein for myelin formation in the CNS and PNS (Alberts et al., 2018).
The PI4KIII-alpha (PI4KA;600286) complex synthesizes phosphatidylinositol 4-phosphate (PI4P), which is localized at the plasma membrane and essential for the formation of oligodendrocytes (Alvarez-Prats et al., 2018). Moreover, one component of the PI4KA complex, FAM126A or FAM126B (HYCC2), has been found to directly bind to TTC7B (620060) through its N-terminal portion. The FAM126A-TTC7B heterodimer is instantaneously bound to PI4KIII-alpha to form a ternary complex (Baskin et al., 2016). Therefore, the PI4KIII-alpha complex plays a regulatory role in PI4P synthesis (Baskin et al., 2016). As previously described, FAM126A plays a key role in forming oligodendrocytes. The interaction of PI4KIII-alpha with TTC7B and FAM126A is responsible for catalytic activity and stabilization of PI4KIII-alpha.
On the other hand, FAM126A dysfunction may affect oligodendrocyte myelination mediated through lipid synthesis and lipid-associated signaling pathways. Transgenic mice expressing the FAM126 mutant were generated as HLD-5 model mice and showed significantly decreased expression of FAM126 in the corpus callosum and abnormal myelination (Torii et al., 2014). Also, the occurrence of mutations in the members of the PI4KIII-alpha complex, such as TTC7B and PI4K, leads to similar diseases (Baple et al., 2022). Consequently, the clinical symptoms and hypomyelination observed in HLD-5 patients are caused by this complex's disruption of myelin production.
HLD-5 manifests with characteristic clinical conditions impacting the eyes, spine, muscles, CNS, PNS, and remarkably hypomyelination with congenital cataracts (HCC). These clinical symptoms are bilateral congenital cataracts, developmental delay, cerebellar ataxia, slowly progressive gait disturbance, and cognitive impairment (Sarret, 2020).
Herein, we report a case of the HLD-5 variant in a 31-year-old male suffering from progressive neurological impairment and congenital cataracts. Whole exome sequencing (WES) revealed a pathogenic homozygous variant in proband, and both parents were carriers of the same genotype. In the present study, we recruited a family from Eastern Iran with a consanguineous marriage and detailed the clinical characteristics of the affected individual. The clinical presentations and the identification of a homozygous variant in the FAM126A gene allowed us to attribute the disorder to HLD-5. So far, 16 different variants have been reported in the FAM126A gene.
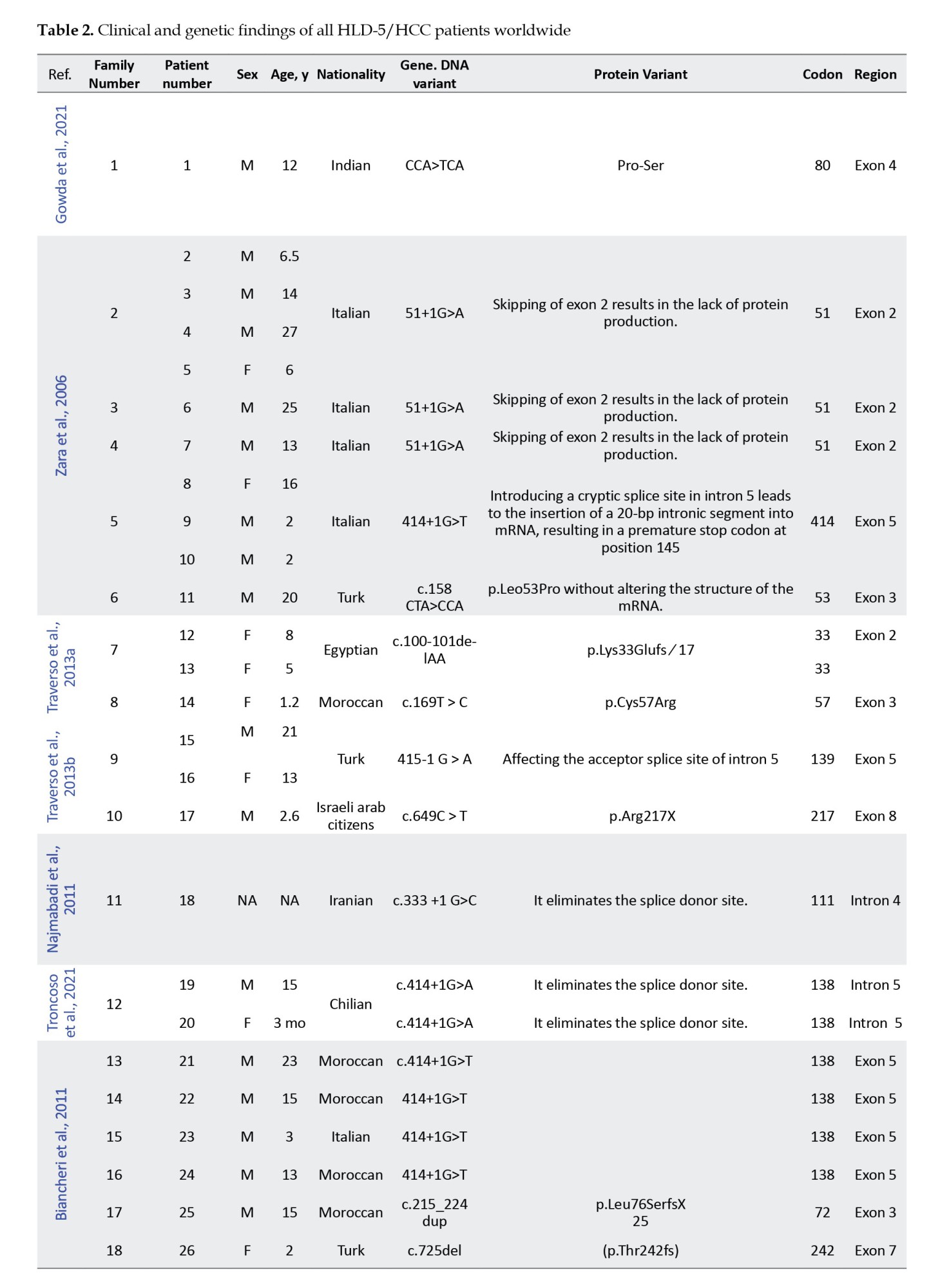
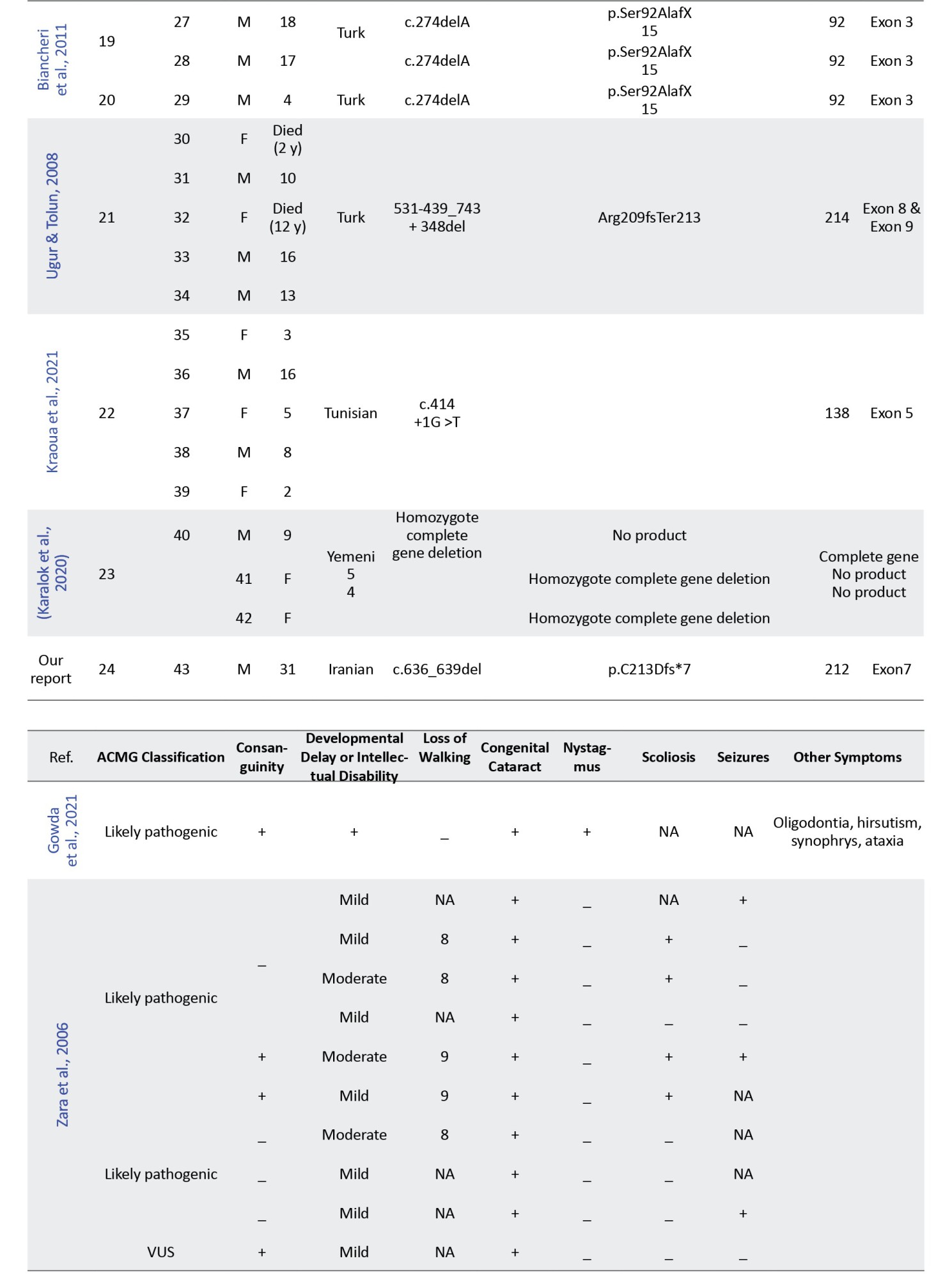
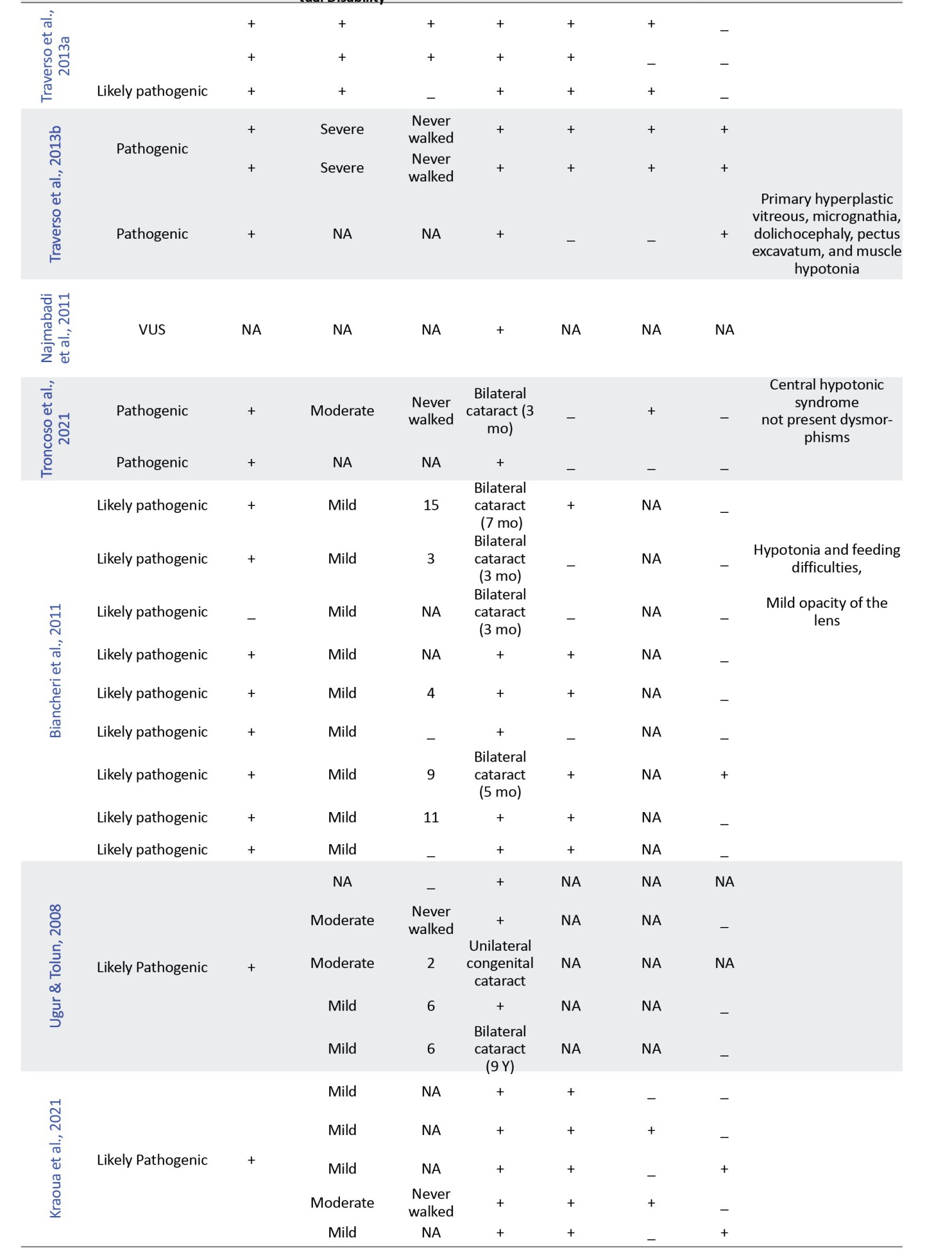
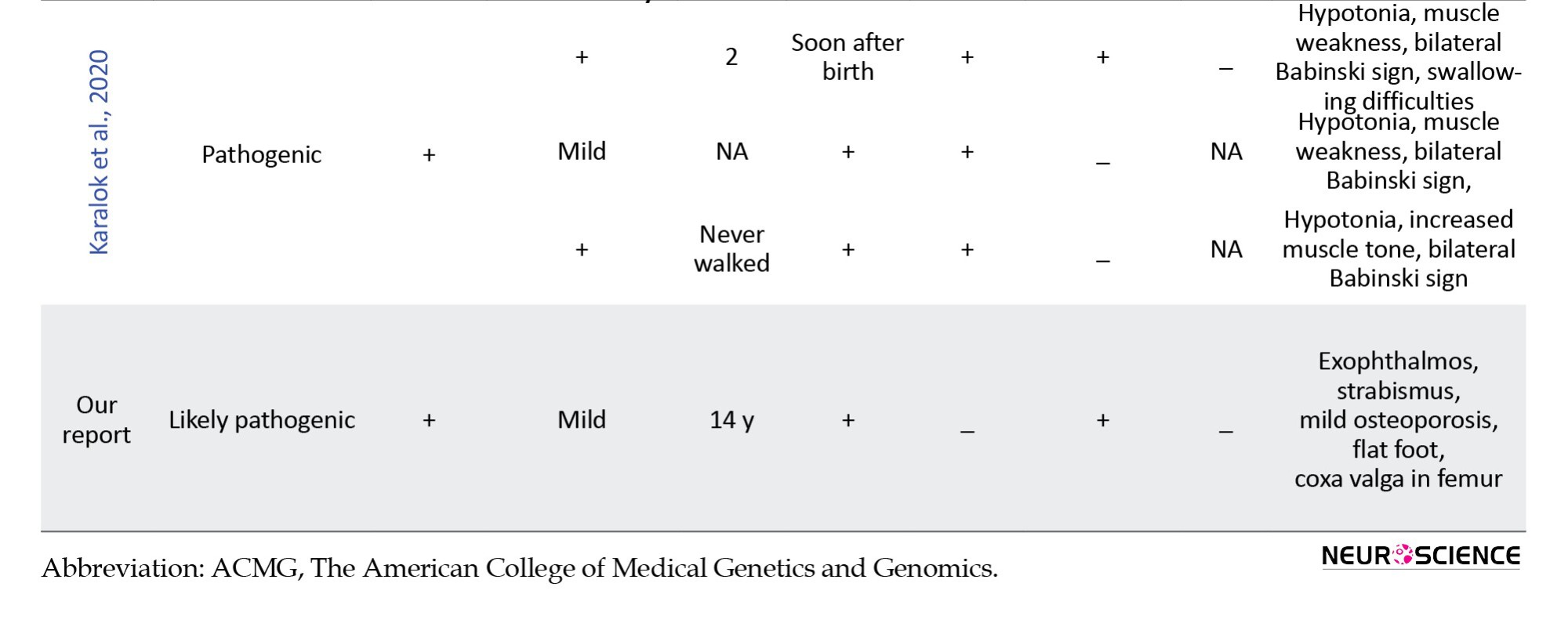
In this study, we retrospectively reviewed our experience with HLD-5 and 42 previously published cases based on the FAM126A gene. We reviewed the clinical and molecular characteristics of all reported patients with the FAM126A mutation and summarized them in Table 2.
Case presentation
The proband was a 31-year-old Iranian man whose parents were double first cousins, and his mother’s relatives had a history of complete paralysis. A neurologist examined the patient. No history of hearing impairment was observed. Milestones of motor development were delayed. He presented with progressive gait disturbance at the age of 14 years. Motor impairments with gait and unsteadiness, as well as difficulties in both climbing up and down the stairs, were also noted.
Magnetic resonance imaging (MRI) of the brain revealed bright signal changes in the centrum semiovale and periventricular area that were compatible with myelin disorders and dysmyelogenesis at 11 years of age. Furthermore, brain MRI studies at age 29 showed moderate to severe diffuse brain atrophy and signal intensity changes in the white matter. Also, the basal ganglia, brain stem, and thalamus show normal signal intensity.
Electromyography (EMG) and nerve conduction velocity (NCV) examination showed increased latency and decreased NCV in the right Median nerve. Also, no response was observed in the stimulation of the right superficial peroneal and anterior tibialis nerves, indicating peripheral neuropathy. Finally, a patient with peripheral neuropathy, loss of ability to walk progressively, delayed motor development, mild Intellectual disability, and congenital cataracts were referred to the neurology service in our study. Both parents were non-symptomatic. We obtained written informed consent from the patient’s legal representatives according to the established ethical protocol guidelines.
2. Materials and Methods
Literature review
A systematic literature search was carried out by searching relevant keywords “HLD-5,” “DRCTNNB1A,” “HYCC1,” and “FAM126A,” as well as relevant MeSH terms in PubMed and Google Scholar electronic databases. The inclusion criteria encompassed studies evaluating HLD-5, focusing on clinical presentation and or molecular genetic testing targeting the FAM126A gene. Studies without HLD-5 or HYCC1 mutation were excluded. Records were screened for eligibility, and data were extracted from the articles included by two independent reviewers. Demographic and clinical features of patients with pathogenic variants of HLD-5 and FAM126A are summarized in Table 2. Afterward, the data were evaluated to provide a broad overview of the reported cases of HLD-5, encompassing both molecular outcomes and clinical manifestations.
Genomic DNA extraction and WES
Genomic DNA was extracted from the venous blood sample taken from the proband and his parents by a salting-out method. Quantitative and qualitative analysis of DNA was performed using a NanoDrop 2000 spectrophotometer. The extracted DNA was fragmented, adapted, barcoded, and subjected to solution phase hybridization, so exome libraries were constructed using Twist Exome 2.0. Then, WES was used to detect exon mutations by the Illumina NovaSeq 6000 sequencing platform. Sequencing data were obtained in FASTQ format, which was converted to BAM files and then variant call format (vcf) files. The Genome Analysis Toolkit (GATK) was used to identify indel and SNVs. The ANNOVAR tool was also used to annotate genetic variant results in detail. Exomes were sequenced to target coverage of 100X. The raw sequence data were aligned to the human genome reference (GRCh37) using the Burrows-Wheeler Aligner (BWA).
We then filtered out variants with an allelic frequency greater than 1% in the databases, including the 1000 Genomes database, the dbSNP, the ExAC03, HapMap, ESP6500, and an in-house database. Candidate variants were classified based on the American College of Medical Genetics (ACMG) guidelines (Richards et al., 2015) and ClinGen specifications (Zhang et al., 2022b). Finally, sorting of intolerance from tolerance (SIFT) and polymorphism phenotyping v2 (PolyPhen-2) analyses were performed to determine the pathogenicity of candidate genes. Only disease-causing or disease-associated genetic variants are reported. PROVEAN and MAPP software were used to identify the structure or function and evolutionary conservation.
Sanger sequencing
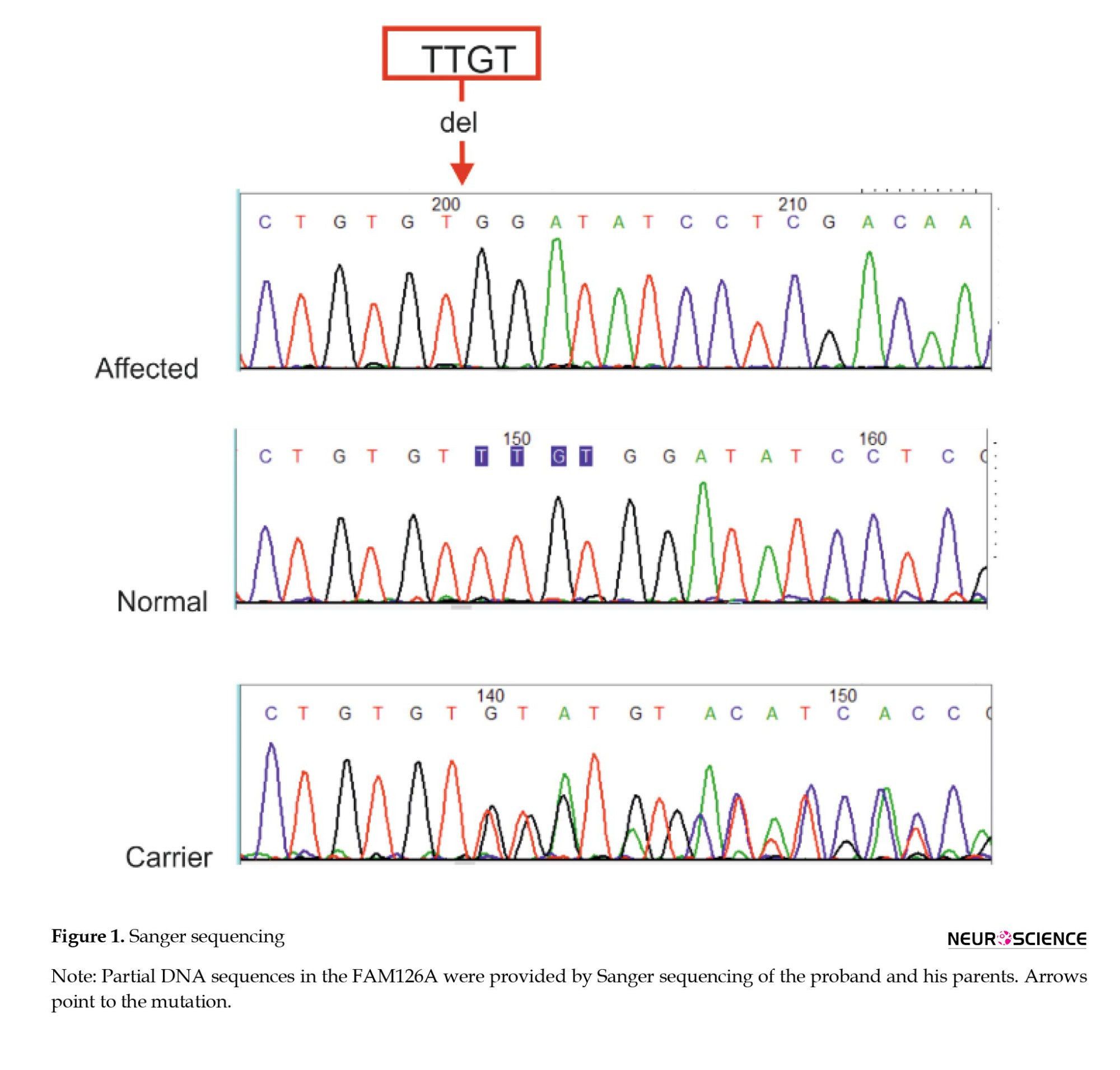
Sanger sequencing was performed to verify the DNA sequence variants identified through exome sequencing within the proband and co-segregation analysis
(Figure 1). PCR amplification was performed using specific primers for the target region using Primer3 online software (Primer3, 2025) corresponding to the FAM126A gene. The sequences of the primers were 5′-TCTGTGTATCAACATACCTCAGCT-3′ and 3′-ACTGATTTTCATTCCGTCCTTGA-5′. PCR products with a size of 596 bp were subjected to direct sequencing by ABI 3730XL DNA Analyzer (ThermoFisher Scientific) capillary sequencing, and the chromas software analyzed the results. According to the Franklin database, the p.C213Dfs*7 substitution is a novel frameshift null variant of the X-chromosomal FAM126A gene. The mentioned variant is predicted to undergo mRNA decay (NMD) because this premature termination codon-causing mutation is located in exon 8, which is not the last exon or the last 50 bp of the preliminary exon. However, it was classified as a likely pathogenic variant according to the ACMG criteria.
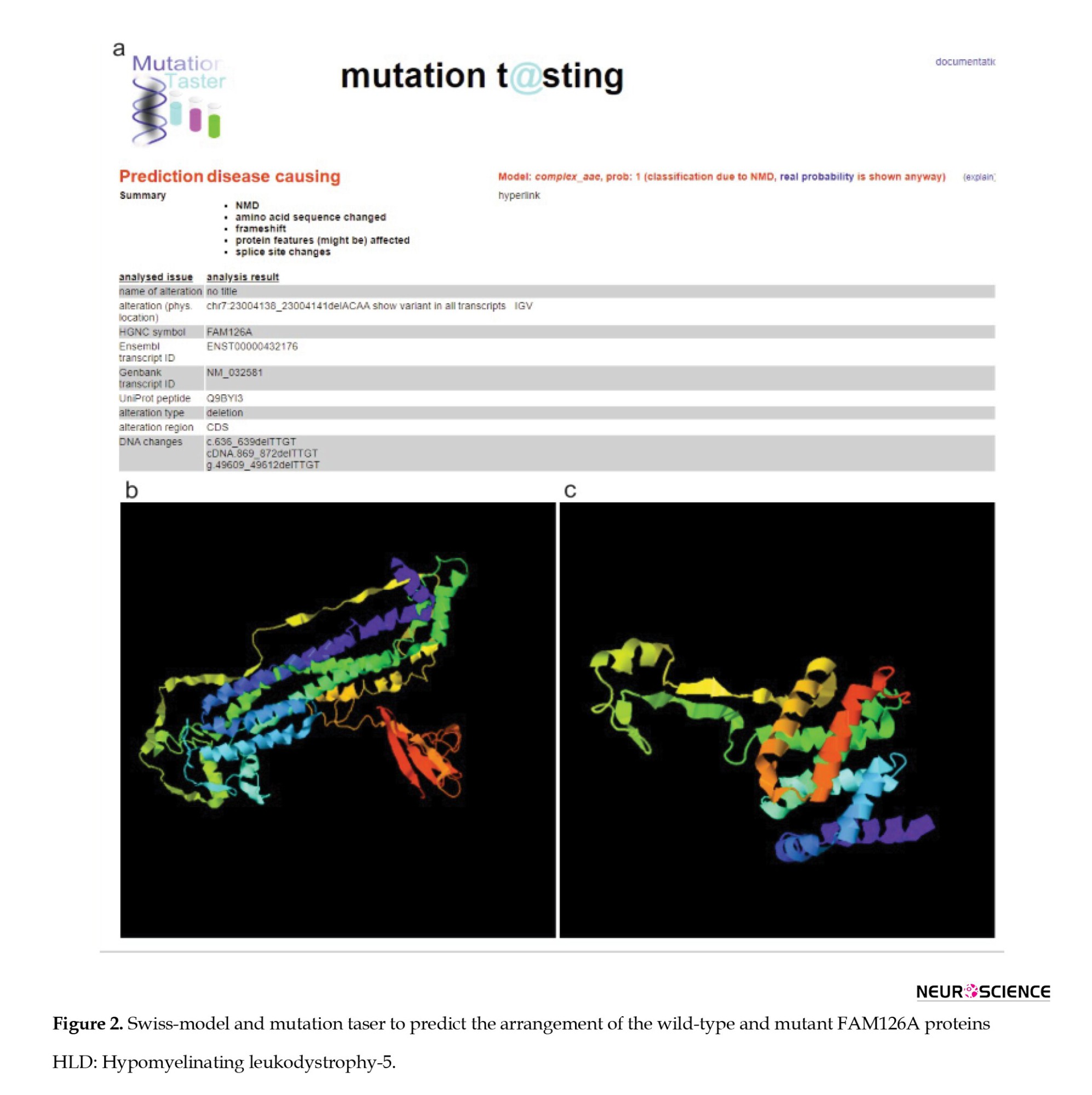
Bioinformatics analysis
We predicted the potential deleterious effects of the novel mutation using Mutation Taster (2025).
Swiss-Model software (2025) made three-dimensional protein structure models (Figure 2).
3. Results
Using WES data analysis, we identified a novel homozygous mutation (NM_032581: c.636_639del p.C213Dfs*7) in the FAM126A gene. This variant causes a shift in the reading frame starting at codon 213, and the patient’s phenotype was consistent with autosomal recessive hypomyelination leukodystrophy type 5. The mentioned variant can cause premature termination of amino acid translation or affect mRNA expression. Sanger sequencing was performed to confirm the presence of the identified variant. Based on current scientific knowledge, the variant is likely pathogenic. According to the ACMG standards for the classification of human gene variants, the p.C213Dfs*7 was identified as PVS1 (predicted null variant in a gene where LOF is a known mechanism of disease and predicted to undergo NMD) and PM2 (absent from controls in GnomAD, Exome Sequencing project, 1000 Genomes, or ExAC) with an ACMG score of 9. Thus, p.C213Dfs*7 was classified as a likely pathogenic variant.
Literature review results
Initially, 47 results were obtained from the PubMed search and 844 results from Google Scholar. Duplicate results were later removed from the search. Articles in languages other than English or non-human studies were excluded based on predetermined criteria. After reviewing the full text of the remaining articles, 10 were included for data extraction. A total of 42 patients with a confirmed molecular diagnosis of HLD-5 or HHC, with homozygous variants in the FAM126A gene, were identified through database analysis (Table 2). Our data indicated that homozygous exonic splicing mutations are the most common pathogenic variant (23 out of 43) reported in the FAM126A gene. The pathological variation c.414+1G>A was the most prevalent mutation detected in 14 out of 43 patients (32.5%). Causative mutations in the FAM126A gene are predominantly protein-truncating (11 of 15 variant types), including frameshift, deletion, and splice site mutations. However, missense variants and whole gene deletions have also been reported.
Common clinical manifestations in the literature and our case were developmental delay or intellectual disability (43/43), loss of walking (23/43), congenital cataract (35/43), nystagmus (20/43), scoliosis (13/43), and seizures (9/43). Patients can have variable clinical manifestations and disease severity, suggesting the effect of modifier genes.
4. Discussion
In the present study, we comprehensively reviewed the literature on FAM126A mutations and HLD-5, focusing on genotype-phenotype correlations. So far, 23 families and 42 patients have been reported. In the present study, we describe an Iranian HCC patient carrying a novel mutation in the FAM126A gene, c.636_639del (p.C213Dfs*7). Examination of the primary amino acid sequence of FAM126A reveals a highly conserved, structured N-terminal domain (residues 1-289) shared by both splice forms and a poorly conserved, disordered C-terminal tail (residues 290-521) (Baskin et al., 2016). Our variant is located in a highly conserved region, leading to disruption of the N-terminal domain of the FAM126A protein and elimination of the C-terminal domain. In addition to the mentioned symptoms, our case presented other rare symptoms such as exophthalmos, strabismus, mild osteoporosis, flat foot, and coxa valga in the femur.
The FAM126A gene comprises 14 exons and codes for a 521 amino acid protein. Hypomyelination and congenital cataract (HCC) is a rare genetic disease caused by pathogenic variants in the FAM126A gene. FAM126A/hyccin regulates phosphatidylinositol 4-phosphate (PI4P) synthesis, contributing to the pool of polyanionic lipids defining plasma membrane identity. Phosphatidylinositol 4-kinase is a three-component complex that has recently been characterized and consists of FAM126A, PI4KIIIα subunit, and its adaptors TTC7 and EFR3B, which are conserved from yeast to mammals (Baskin et al., 2016). This complex is anchored to the PM by a transmembrane protein EFR3, the mammalian homolog of yeast Efr3, which is crucial for recruiting PI4KIIIα to the PM. However, TTC7, the mammalian homolog of yeast Ypp1, is a shuttling protein and binds to EFR3 and PI4KA (Ulengin-Talkish et al., 2021). Finally, FAM126A, present only in higher eukaryotes, and TTC7 form a tight heterodimer with a large protein-protein interface and a conserved surface that increases PM recruitment of PI4KIIIα (Baskin et al., 2016; Ulengin-Talkish et al., 2021).
PI4KA/TTC7/FAM126A heterotrimers probably promote PI4KA activity by stabilizing and orienting its active site toward the membrane. Additionally, the irregular C-terminus of FAM126A protein controls the PI4KA catalytic activity in vitro through an unknown mechanism. This implies that the localization, assembly, and activity of the PI4KA complex is tightly controlled, but the specific pathway involved in this process in mammals remains elusive. Without the FAM126A gene, a general destabilization and degradation of the PI4KIIIα complex components occurs (Ulengin-Talkish et al., 2021).
Sequence variations in TTC7 and FAM126A genes are associated with a heterogeneous group of neurological (FAM126A) and immunological (TTC7A) disorders. Furthermore, biallelic variants in the PI4KA sequence are related to neurological diseases, especially HLD. Comparable organ-specific diseases, such as HLD and HCC, have been described in patients with FAM126A genes pathogenic variants. It is conceivable that the mentioned phenotypical outcomes arise from deficiencies in the specific functions of the PI4KIIIα-TTC7-FAM126 complex due to alterations in catalytic activity or intra-complex functional interactions (Salter et al., 2021).
More than 10 mutations in the FAM126A gene have been described in patients with HLD-5/HCC, most of which are splicing, missense/nonsense, small deletions, small insertions, and gross deletions. Consistently, fibroblast cells from HCC patients with nonsense or missense mutations in the FAM126A gene had significantly reduced expression of PI4KIIIa, PI4P, EFR3A, TTC7A, and TTC7B (Zhang et al., 2022). Thus, HCC has a common pathogenesis involving a disturbance in PI4P production in oligodendrocytes, whose specific function involves massive plasma membrane expansion and, therefore, the generation of PI4P and downstream phosphoinositides (Baskin et al., 2016).
Over 15 FAM126A variants have been identified in diverse geographical populations, including Morocco, Turkey, Israel, Iran, India, Italy, Egypt, Chile, Tunisia, and Yemen. Therefore, HLD/HCC is common (81%) in some countries of the Mediterranean region, which tend to have high rates of consanguinity. HLD/HCC primarily affects men (28 out of 43 cases). Differences in clinical phenotypes have been reported among unrelated individuals sharing the same alteration. For example, out of 9 probands carrying the c.414+1G>T mutation, three did not show congenital cataracts (Biancheri et al., 2011; Kraoua et al., 2021; Zara et al., 2006).
Similarly, probands with a large intragenic deletion involving 2 exons (c.531-439_743+348 del) developed cataracts at birth or in the first 9 years of life (Ugur & Tolun, 2008). Mutations affecting intron 4 (c.415-1 G>A) were associated with a severe phenotype (Traverso et al., 2013). In contrast, mutations affecting c.414+1G>A were identified in probands with milder phenotypes (Troncoso et al., 2021). The significant clinical variability suggests that other genetic or environmental factors likely modify the HLD/HCC phenotype (Biancheri et al., 2011).
Mutations affecting mRNA splicing are the most common molecular findings in patients with HLD-5. These variants are expected to significantly disrupt the acceptor/donor splice sites, leading to exon skipping or the introduction of cryptic splice sites, resulting in a lack of protein production (Table 2).
Although most FAM126A variants lead to disruption of pre-mRNA splicing and consequent changes in protein conformation and activity, the Leu-53-to-Pro mutation can still produce some hyccin protein. However, this variant can accumulate misfolded proteins in the endoplasmic reticulum (ER). In contrast, the wild-type hyccin protein mainly localizes in the cytoplasm. It is plausible that HLD-5-associated variants of FAM126A lead to a decline in the localization of FAM126A protein in plasma membranes (Miyamoto et al., 2014).
On the other hand, Traverso (Traverso et al., 2013) reported that patients with the p.Cys57Arg variant showed a 40% reduction in hyccin protein levels, while patients with the p.Leo53Pro variant experienced an approximately 70% decrease (Traverso et al., 2013). Additionally, cells of patients with nonsense variants such as p.Arg217X completely lacked hyccin protein. Furthermore, reintroducing GFP-tagged FAM126A into patient fibroblasts using recombinant lentivirus as a gene therapy vector partially rescues the phenotypes. All reported deletion variants, including c.100-101delAA, c.725del, c.274delA, 531-439_743+348del, and c.636_639del in FAM126A gene, cause frameshift mutations and create premature stop codons that it significantly disrupts the protein structure. These variants were in exons 2, 3, 7, 8, and 9. Cataracts are not invariably congenital or necessary for diagnosing HCC and can be seen later in life. Moreover, the onset of neurological conditions varied among cases (Baskin et al., 2016; Karalok et al., 2020). Therefore, even in the absence of cataracts or with atypical clinical manifestations, increased T2 signal from the pyramidal tracts in the mesencephalon and pons suggests the diagnosis of HCC and molecular analysis of FAM126A should be the first diagnostic step (Baskin et al., 2016).
5. Conclusion
In this study, clinical manifestations and molecular findings of HLD-5/HCC were explained. Additionally, we reported a novel variant and some clinical features, such as exophthalmos and strabismus, in our proband for the first time. This broadens the pathologic spectrum in HLD-5/HCC patients and highlights the pathologic and clinical variability associated with the same genetic variation, suggesting an essential role for modifier genes in the pathogenesis of HLD-5/HCC.
Ethical Considerations
Compliance with ethical guidelines
Ethical approval was issued by the Ethics Committee of Isfahan University of Medical Sciences, Isfahan, Iran (Code: IR.MUI.MED.REC.1402.398) for this study. Written informed consent was obtained from the patient’s legal guardians to publish this manuscript and any accompanying images.
Funding
This article is based on a portion of the PhD dissertation of Sahar Bayat, approved by the Department of Genetics and Molecular Biology, Faculty of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran. This study was supported by Isfahan University of Medical Sciences, Isfahan, Iran (Project code: 60757).
Authors' contributions
Conceptualization, study design, review, editing and final approval: All authors; Material preparation, data collection and analysis: Sahar Bayat, Milad Gholami, Majid Kheirollahi, Mohammadreza Ghazavi, Hamidreza Khodadadi and Jafar Nasiri; Writing the original draft: Sahar Bayat.
Conflict of interest
The authors declared no conflict of interest."
Acknowledgments
The authors appreciate the participation of patients and individuals in this study.
References
Myelin sheath around axons is formed by the spiral wrapping of the differentiated oligodendroglial cell plasma membrane in the central nervous system (CNS) and Schwann cell plasma membrane in the peripheral nervous system (PNS) (Simons & Nave, 2016). These myelin sheaths play an intricate role in saltatory conduction and protecting axons from cellular stresses (Nave & Werner, 2021). Hypomyelinating leukodystrophy (HLD) is a group of genetic demyelinating or dysmyelinating diseases that affect the proper development of the myelin sheath in the CNS (Charzewska et al., 2016).
HLDs are generally rare, heterogeneous, and challenging to diagnose because they have diverse clinical manifestations that most often include neurological dysfunction such as ataxia, motor ability, and intellectual disability (Charzewska et al., 2016). Next-generation sequencing (NGS) has revolutionized the potential to identify multiple autosomal recessive HLD genes (Torii & Yamauchi, 2023).
According to the Online Mendelian Inheritance in Man (OMIM, 2025), the number of HLDs and responsible genes identified in the last 10 years has increased (Table 1).
Among HLDs, HLD-5 is a rare genetic disorder caused by an autosomal recessive mutation in the FAM126A (formerly DRCTNNB1A) gene (Torii & Yamauchi, 2023). The FAM126A gene, also known as hyccin PI4KA lipid kinase complex subunit 1 (HYCC1), is located on chromosome 7p15.3, which contains 14 exons and 13 introns. This gene encodes a protein containing 521 amino acids (National Library of Medicine, 2025).
FAM126A expression is downregulated by beta-catenin, an essential protein for myelin formation in the CNS and PNS (Alberts et al., 2018).
The PI4KIII-alpha (PI4KA;600286) complex synthesizes phosphatidylinositol 4-phosphate (PI4P), which is localized at the plasma membrane and essential for the formation of oligodendrocytes (Alvarez-Prats et al., 2018). Moreover, one component of the PI4KA complex, FAM126A or FAM126B (HYCC2), has been found to directly bind to TTC7B (620060) through its N-terminal portion. The FAM126A-TTC7B heterodimer is instantaneously bound to PI4KIII-alpha to form a ternary complex (Baskin et al., 2016). Therefore, the PI4KIII-alpha complex plays a regulatory role in PI4P synthesis (Baskin et al., 2016). As previously described, FAM126A plays a key role in forming oligodendrocytes. The interaction of PI4KIII-alpha with TTC7B and FAM126A is responsible for catalytic activity and stabilization of PI4KIII-alpha.
On the other hand, FAM126A dysfunction may affect oligodendrocyte myelination mediated through lipid synthesis and lipid-associated signaling pathways. Transgenic mice expressing the FAM126 mutant were generated as HLD-5 model mice and showed significantly decreased expression of FAM126 in the corpus callosum and abnormal myelination (Torii et al., 2014). Also, the occurrence of mutations in the members of the PI4KIII-alpha complex, such as TTC7B and PI4K, leads to similar diseases (Baple et al., 2022). Consequently, the clinical symptoms and hypomyelination observed in HLD-5 patients are caused by this complex's disruption of myelin production.
HLD-5 manifests with characteristic clinical conditions impacting the eyes, spine, muscles, CNS, PNS, and remarkably hypomyelination with congenital cataracts (HCC). These clinical symptoms are bilateral congenital cataracts, developmental delay, cerebellar ataxia, slowly progressive gait disturbance, and cognitive impairment (Sarret, 2020).
Herein, we report a case of the HLD-5 variant in a 31-year-old male suffering from progressive neurological impairment and congenital cataracts. Whole exome sequencing (WES) revealed a pathogenic homozygous variant in proband, and both parents were carriers of the same genotype. In the present study, we recruited a family from Eastern Iran with a consanguineous marriage and detailed the clinical characteristics of the affected individual. The clinical presentations and the identification of a homozygous variant in the FAM126A gene allowed us to attribute the disorder to HLD-5. So far, 16 different variants have been reported in the FAM126A gene.
In this study, we retrospectively reviewed our experience with HLD-5 and 42 previously published cases based on the FAM126A gene. We reviewed the clinical and molecular characteristics of all reported patients with the FAM126A mutation and summarized them in Table 2.
Case presentation
The proband was a 31-year-old Iranian man whose parents were double first cousins, and his mother’s relatives had a history of complete paralysis. A neurologist examined the patient. No history of hearing impairment was observed. Milestones of motor development were delayed. He presented with progressive gait disturbance at the age of 14 years. Motor impairments with gait and unsteadiness, as well as difficulties in both climbing up and down the stairs, were also noted.
Magnetic resonance imaging (MRI) of the brain revealed bright signal changes in the centrum semiovale and periventricular area that were compatible with myelin disorders and dysmyelogenesis at 11 years of age. Furthermore, brain MRI studies at age 29 showed moderate to severe diffuse brain atrophy and signal intensity changes in the white matter. Also, the basal ganglia, brain stem, and thalamus show normal signal intensity.
Electromyography (EMG) and nerve conduction velocity (NCV) examination showed increased latency and decreased NCV in the right Median nerve. Also, no response was observed in the stimulation of the right superficial peroneal and anterior tibialis nerves, indicating peripheral neuropathy. Finally, a patient with peripheral neuropathy, loss of ability to walk progressively, delayed motor development, mild Intellectual disability, and congenital cataracts were referred to the neurology service in our study. Both parents were non-symptomatic. We obtained written informed consent from the patient’s legal representatives according to the established ethical protocol guidelines.
2. Materials and Methods
Literature review
A systematic literature search was carried out by searching relevant keywords “HLD-5,” “DRCTNNB1A,” “HYCC1,” and “FAM126A,” as well as relevant MeSH terms in PubMed and Google Scholar electronic databases. The inclusion criteria encompassed studies evaluating HLD-5, focusing on clinical presentation and or molecular genetic testing targeting the FAM126A gene. Studies without HLD-5 or HYCC1 mutation were excluded. Records were screened for eligibility, and data were extracted from the articles included by two independent reviewers. Demographic and clinical features of patients with pathogenic variants of HLD-5 and FAM126A are summarized in Table 2. Afterward, the data were evaluated to provide a broad overview of the reported cases of HLD-5, encompassing both molecular outcomes and clinical manifestations.
Genomic DNA extraction and WES
Genomic DNA was extracted from the venous blood sample taken from the proband and his parents by a salting-out method. Quantitative and qualitative analysis of DNA was performed using a NanoDrop 2000 spectrophotometer. The extracted DNA was fragmented, adapted, barcoded, and subjected to solution phase hybridization, so exome libraries were constructed using Twist Exome 2.0. Then, WES was used to detect exon mutations by the Illumina NovaSeq 6000 sequencing platform. Sequencing data were obtained in FASTQ format, which was converted to BAM files and then variant call format (vcf) files. The Genome Analysis Toolkit (GATK) was used to identify indel and SNVs. The ANNOVAR tool was also used to annotate genetic variant results in detail. Exomes were sequenced to target coverage of 100X. The raw sequence data were aligned to the human genome reference (GRCh37) using the Burrows-Wheeler Aligner (BWA).
We then filtered out variants with an allelic frequency greater than 1% in the databases, including the 1000 Genomes database, the dbSNP, the ExAC03, HapMap, ESP6500, and an in-house database. Candidate variants were classified based on the American College of Medical Genetics (ACMG) guidelines (Richards et al., 2015) and ClinGen specifications (Zhang et al., 2022b). Finally, sorting of intolerance from tolerance (SIFT) and polymorphism phenotyping v2 (PolyPhen-2) analyses were performed to determine the pathogenicity of candidate genes. Only disease-causing or disease-associated genetic variants are reported. PROVEAN and MAPP software were used to identify the structure or function and evolutionary conservation.
Sanger sequencing
Sanger sequencing was performed to verify the DNA sequence variants identified through exome sequencing within the proband and co-segregation analysis
(Figure 1). PCR amplification was performed using specific primers for the target region using Primer3 online software (Primer3, 2025) corresponding to the FAM126A gene. The sequences of the primers were 5′-TCTGTGTATCAACATACCTCAGCT-3′ and 3′-ACTGATTTTCATTCCGTCCTTGA-5′. PCR products with a size of 596 bp were subjected to direct sequencing by ABI 3730XL DNA Analyzer (ThermoFisher Scientific) capillary sequencing, and the chromas software analyzed the results. According to the Franklin database, the p.C213Dfs*7 substitution is a novel frameshift null variant of the X-chromosomal FAM126A gene. The mentioned variant is predicted to undergo mRNA decay (NMD) because this premature termination codon-causing mutation is located in exon 8, which is not the last exon or the last 50 bp of the preliminary exon. However, it was classified as a likely pathogenic variant according to the ACMG criteria.
Bioinformatics analysis
We predicted the potential deleterious effects of the novel mutation using Mutation Taster (2025).
Swiss-Model software (2025) made three-dimensional protein structure models (Figure 2).
3. Results
Using WES data analysis, we identified a novel homozygous mutation (NM_032581: c.636_639del p.C213Dfs*7) in the FAM126A gene. This variant causes a shift in the reading frame starting at codon 213, and the patient’s phenotype was consistent with autosomal recessive hypomyelination leukodystrophy type 5. The mentioned variant can cause premature termination of amino acid translation or affect mRNA expression. Sanger sequencing was performed to confirm the presence of the identified variant. Based on current scientific knowledge, the variant is likely pathogenic. According to the ACMG standards for the classification of human gene variants, the p.C213Dfs*7 was identified as PVS1 (predicted null variant in a gene where LOF is a known mechanism of disease and predicted to undergo NMD) and PM2 (absent from controls in GnomAD, Exome Sequencing project, 1000 Genomes, or ExAC) with an ACMG score of 9. Thus, p.C213Dfs*7 was classified as a likely pathogenic variant.
Literature review results
Initially, 47 results were obtained from the PubMed search and 844 results from Google Scholar. Duplicate results were later removed from the search. Articles in languages other than English or non-human studies were excluded based on predetermined criteria. After reviewing the full text of the remaining articles, 10 were included for data extraction. A total of 42 patients with a confirmed molecular diagnosis of HLD-5 or HHC, with homozygous variants in the FAM126A gene, were identified through database analysis (Table 2). Our data indicated that homozygous exonic splicing mutations are the most common pathogenic variant (23 out of 43) reported in the FAM126A gene. The pathological variation c.414+1G>A was the most prevalent mutation detected in 14 out of 43 patients (32.5%). Causative mutations in the FAM126A gene are predominantly protein-truncating (11 of 15 variant types), including frameshift, deletion, and splice site mutations. However, missense variants and whole gene deletions have also been reported.
Common clinical manifestations in the literature and our case were developmental delay or intellectual disability (43/43), loss of walking (23/43), congenital cataract (35/43), nystagmus (20/43), scoliosis (13/43), and seizures (9/43). Patients can have variable clinical manifestations and disease severity, suggesting the effect of modifier genes.
4. Discussion
In the present study, we comprehensively reviewed the literature on FAM126A mutations and HLD-5, focusing on genotype-phenotype correlations. So far, 23 families and 42 patients have been reported. In the present study, we describe an Iranian HCC patient carrying a novel mutation in the FAM126A gene, c.636_639del (p.C213Dfs*7). Examination of the primary amino acid sequence of FAM126A reveals a highly conserved, structured N-terminal domain (residues 1-289) shared by both splice forms and a poorly conserved, disordered C-terminal tail (residues 290-521) (Baskin et al., 2016). Our variant is located in a highly conserved region, leading to disruption of the N-terminal domain of the FAM126A protein and elimination of the C-terminal domain. In addition to the mentioned symptoms, our case presented other rare symptoms such as exophthalmos, strabismus, mild osteoporosis, flat foot, and coxa valga in the femur.
The FAM126A gene comprises 14 exons and codes for a 521 amino acid protein. Hypomyelination and congenital cataract (HCC) is a rare genetic disease caused by pathogenic variants in the FAM126A gene. FAM126A/hyccin regulates phosphatidylinositol 4-phosphate (PI4P) synthesis, contributing to the pool of polyanionic lipids defining plasma membrane identity. Phosphatidylinositol 4-kinase is a three-component complex that has recently been characterized and consists of FAM126A, PI4KIIIα subunit, and its adaptors TTC7 and EFR3B, which are conserved from yeast to mammals (Baskin et al., 2016). This complex is anchored to the PM by a transmembrane protein EFR3, the mammalian homolog of yeast Efr3, which is crucial for recruiting PI4KIIIα to the PM. However, TTC7, the mammalian homolog of yeast Ypp1, is a shuttling protein and binds to EFR3 and PI4KA (Ulengin-Talkish et al., 2021). Finally, FAM126A, present only in higher eukaryotes, and TTC7 form a tight heterodimer with a large protein-protein interface and a conserved surface that increases PM recruitment of PI4KIIIα (Baskin et al., 2016; Ulengin-Talkish et al., 2021).
PI4KA/TTC7/FAM126A heterotrimers probably promote PI4KA activity by stabilizing and orienting its active site toward the membrane. Additionally, the irregular C-terminus of FAM126A protein controls the PI4KA catalytic activity in vitro through an unknown mechanism. This implies that the localization, assembly, and activity of the PI4KA complex is tightly controlled, but the specific pathway involved in this process in mammals remains elusive. Without the FAM126A gene, a general destabilization and degradation of the PI4KIIIα complex components occurs (Ulengin-Talkish et al., 2021).
Sequence variations in TTC7 and FAM126A genes are associated with a heterogeneous group of neurological (FAM126A) and immunological (TTC7A) disorders. Furthermore, biallelic variants in the PI4KA sequence are related to neurological diseases, especially HLD. Comparable organ-specific diseases, such as HLD and HCC, have been described in patients with FAM126A genes pathogenic variants. It is conceivable that the mentioned phenotypical outcomes arise from deficiencies in the specific functions of the PI4KIIIα-TTC7-FAM126 complex due to alterations in catalytic activity or intra-complex functional interactions (Salter et al., 2021).
More than 10 mutations in the FAM126A gene have been described in patients with HLD-5/HCC, most of which are splicing, missense/nonsense, small deletions, small insertions, and gross deletions. Consistently, fibroblast cells from HCC patients with nonsense or missense mutations in the FAM126A gene had significantly reduced expression of PI4KIIIa, PI4P, EFR3A, TTC7A, and TTC7B (Zhang et al., 2022). Thus, HCC has a common pathogenesis involving a disturbance in PI4P production in oligodendrocytes, whose specific function involves massive plasma membrane expansion and, therefore, the generation of PI4P and downstream phosphoinositides (Baskin et al., 2016).
Over 15 FAM126A variants have been identified in diverse geographical populations, including Morocco, Turkey, Israel, Iran, India, Italy, Egypt, Chile, Tunisia, and Yemen. Therefore, HLD/HCC is common (81%) in some countries of the Mediterranean region, which tend to have high rates of consanguinity. HLD/HCC primarily affects men (28 out of 43 cases). Differences in clinical phenotypes have been reported among unrelated individuals sharing the same alteration. For example, out of 9 probands carrying the c.414+1G>T mutation, three did not show congenital cataracts (Biancheri et al., 2011; Kraoua et al., 2021; Zara et al., 2006).
Similarly, probands with a large intragenic deletion involving 2 exons (c.531-439_743+348 del) developed cataracts at birth or in the first 9 years of life (Ugur & Tolun, 2008). Mutations affecting intron 4 (c.415-1 G>A) were associated with a severe phenotype (Traverso et al., 2013). In contrast, mutations affecting c.414+1G>A were identified in probands with milder phenotypes (Troncoso et al., 2021). The significant clinical variability suggests that other genetic or environmental factors likely modify the HLD/HCC phenotype (Biancheri et al., 2011).
Mutations affecting mRNA splicing are the most common molecular findings in patients with HLD-5. These variants are expected to significantly disrupt the acceptor/donor splice sites, leading to exon skipping or the introduction of cryptic splice sites, resulting in a lack of protein production (Table 2).
Although most FAM126A variants lead to disruption of pre-mRNA splicing and consequent changes in protein conformation and activity, the Leu-53-to-Pro mutation can still produce some hyccin protein. However, this variant can accumulate misfolded proteins in the endoplasmic reticulum (ER). In contrast, the wild-type hyccin protein mainly localizes in the cytoplasm. It is plausible that HLD-5-associated variants of FAM126A lead to a decline in the localization of FAM126A protein in plasma membranes (Miyamoto et al., 2014).
On the other hand, Traverso (Traverso et al., 2013) reported that patients with the p.Cys57Arg variant showed a 40% reduction in hyccin protein levels, while patients with the p.Leo53Pro variant experienced an approximately 70% decrease (Traverso et al., 2013). Additionally, cells of patients with nonsense variants such as p.Arg217X completely lacked hyccin protein. Furthermore, reintroducing GFP-tagged FAM126A into patient fibroblasts using recombinant lentivirus as a gene therapy vector partially rescues the phenotypes. All reported deletion variants, including c.100-101delAA, c.725del, c.274delA, 531-439_743+348del, and c.636_639del in FAM126A gene, cause frameshift mutations and create premature stop codons that it significantly disrupts the protein structure. These variants were in exons 2, 3, 7, 8, and 9. Cataracts are not invariably congenital or necessary for diagnosing HCC and can be seen later in life. Moreover, the onset of neurological conditions varied among cases (Baskin et al., 2016; Karalok et al., 2020). Therefore, even in the absence of cataracts or with atypical clinical manifestations, increased T2 signal from the pyramidal tracts in the mesencephalon and pons suggests the diagnosis of HCC and molecular analysis of FAM126A should be the first diagnostic step (Baskin et al., 2016).
5. Conclusion
In this study, clinical manifestations and molecular findings of HLD-5/HCC were explained. Additionally, we reported a novel variant and some clinical features, such as exophthalmos and strabismus, in our proband for the first time. This broadens the pathologic spectrum in HLD-5/HCC patients and highlights the pathologic and clinical variability associated with the same genetic variation, suggesting an essential role for modifier genes in the pathogenesis of HLD-5/HCC.
Ethical Considerations
Compliance with ethical guidelines
Ethical approval was issued by the Ethics Committee of Isfahan University of Medical Sciences, Isfahan, Iran (Code: IR.MUI.MED.REC.1402.398) for this study. Written informed consent was obtained from the patient’s legal guardians to publish this manuscript and any accompanying images.
Funding
This article is based on a portion of the PhD dissertation of Sahar Bayat, approved by the Department of Genetics and Molecular Biology, Faculty of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran. This study was supported by Isfahan University of Medical Sciences, Isfahan, Iran (Project code: 60757).
Authors' contributions
Conceptualization, study design, review, editing and final approval: All authors; Material preparation, data collection and analysis: Sahar Bayat, Milad Gholami, Majid Kheirollahi, Mohammadreza Ghazavi, Hamidreza Khodadadi and Jafar Nasiri; Writing the original draft: Sahar Bayat.
Conflict of interest
The authors declared no conflict of interest."
Acknowledgments
The authors appreciate the participation of patients and individuals in this study.
References
Alberts, R., Visschedijk, M. C., Mucha, S., Ellinghaus, D., Bergquist, A., & Karlsen, H.,.et al. (2018). Low-frequency and rare DNA sequence variants associated with primary sclerosing cholangitis susceptibility. In New genes, rare variants & moving towards clinical practice. Groningen: University of Groningen. [Link]
Alvarez-Prats, A., Bjelobaba, I., Aldworth, Z., Baba, T., Abebe, D., & Kim, Y. J., et al. (2018). Schwann-cell-specific deletion of phosphatidylinositol 4-Kinase Alpha Causes Aberrant Myelination. Cell Reports, 23(10), 2881–2890. [DOI:10.1016/j.celrep.2018.05.019] [PMID]
Baple, E. L., Salter, C., Uhlig, H., Wolf, N. I., & Crosby, A. H. (2022). PI4KA-Related Disorder. In M. P. Adam (Eds.) et. al., GeneReviews®. University of Washington, Seattle. [PMID]
Baskin, J. M., Wu, X., Christiano, R., Oh, M. S., Schauder, C. M., & Gazzerro, E., et al. (2016). The leukodystrophy protein FAM126A (hyccin) regulates PtdIns (4) P synthesis at the plasma membrane. Nature Cell Biology, 18(1), 132-138. [DOI:10.1038/ncb3271] [PMID]
Biancheri, R., Zara, F., Rossi, A., Mathot, M., Nassogne, M. C., &Yalcinkaya, C., et al. (2011). Hypomyelination and congenital cataract: Broadening the clinical phenotype. Archives of Neurology, 68(9), 1191-1194. [DOI:10.1001/archneurol.2011.201] [PMID]
Charzewska, A., Wierzba, J., Iżycka-Świeszewska, E., Bekiesińska-Figatowska, M., Jurek, M., & Gintowt, A., et al. (2016). Hypomyelinating leukodystrophies-a molecular insight into the white matter pathology. Clinical Genetics, 90(4), 293-304. [DOI:10.1111/cge.12811] [PMID]
Eno, C. C., Graakjaer, J., Svaneby, D., Nizon, M., Kianmahd, J., & Signer, R., et al. (2021). 14q32.11 microdeletion including CALM1, TTC7B, PSMC1, and RPS6KA5: A new potential cause of developmental and language delay in three unrelated patients. American Journal of Medical Genetics. Part A, 185(5), 1519–1524.[DOI:10.1002/ajmg.a.62117] [PMID]
Gowda, V. K., Nagarajan, B., Srinivasan, V. M., & Bhat, M. (2021). Expanding phenotype of Hypomyelination and Congenital Cataract (HCC) with a Novel Pathogenic Variant. Indian Journal of Pediatrics, 88(3), 312–313. [DOI:10.1007/s12098-020-03253-8] [PMID]
Karalok, Z. S., Gurkasb, E., Aydinc, K., & Ceylaner, S. (2020). Hypomyelination and Congenital Cataract: Three Siblings Presentation. Journal of Pediatric Neurosciences, 15(3), 270–273. [DOI:10.4103/jpn.JPN_161_18] [PMID]
Kraoua, I., Bouyacoub, Y., Drissi, C., Chargui, M., Rebai, I., & Chebil, A., et al. (2021). Hypomyelination and congenital cataract: Clinical, imaging, and genetic findings in three tunisian families and literature Review. Neuropediatrics, 52(4), 302–309. [DOI:10.1055/s-0041-1728654] [PMID]
Miyamoto, Y., Torii, T., Eguchi, T., Nakamura, K., Tanoue, A., & Yamauchi, J. (2014). Hypomyelinating leukodystrophy-associated missense mutant of FAM126A/hyccin/DRCTNNB1A aggregates in the endoplasmic reticulum. Journal of Clinical Neuroscience: Official Journal of the Neurosurgical Society of Australasia, 21(6), 1033–1039. [DOI:10.1016/j.jocn.2013.09.014] [PMID]
Mutation Taster. (2025). Mutation t@sting. Retrieved from: [Link]
Najmabadi, H., Hu, H., Garshasbi, M., Zemojtel, T., Abedini, S. S., &Chen, W., et al. (2011). Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature, 478(7367), 57–63. [PMID]
Nave, K. A., & Werner, H. B. (2021). Ensheathment and myelination of axons: evolution of glial functions. Annual Review of Neuroscience, 44, 197-219. [DOI:10.1146/annurev-neuro-100120-122621] [PMID]
National Library of Medicine (NCBI). (2025). HYCC1 hyccin PI4KA lipid kinase complex subunit 1 [Homo sapiens (human)]. Retrieved from: [Link]
OMIM. (2025). Number of HLDs and responsible genes. Retrieved from: [Link]
Primer3. (2025). Primer3 (v. 0.4.0) Pick primers from a DNA sequence. Retrieved from: [Link]
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., & Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine : Official Journal of the American College of Medical Genetics, 17(5), 405–424. [DOI:10.1038/gim.2015.30] [PMID]
Salter, C. G., Cai, Y., Lo, B., Helman, G., Taylor, H., & McCartney, A., et al. (2021). Biallelic PI4KA variants cause neurological, intestinal and immunological disease. Brain, 144(12), 3597-3610. [DOI:10.1093/brain/awab313] [PMID]
Sarret, C. (2020). Leukodystrophies and genetic leukoencephalopathies in children. Revue Neurologique, 176(1-2), 10-19. [DOI:10.1016/j.neurol.2019.04.003] [PMID]
Simons, M., & Nave, K. A. (2016). Oligodendrocytes: Myelination and axonal support. Cold Spring Harbor Perspectives in Biology, 8(1), a020479. [DOI:10.1101/cshperspect.a020479] [PMID]
Swiss Model. (2025). Swiss-Model software. Retrieved from: [Link]
Torii, T., Miyamoto, Y., Yamauchi, J., & Tanoue, A. (2014). Pelizaeus-Merzbacher disease: Cellular pathogenesis and pharmacologic therapy. Pediatrics International : Official Journal of the Japan Pediatric Society, 56(5), 659–666. [DOI:10.1111/ped.12450] [PMID]
Torii, T., & Yamauchi, J. (2023). Molecular pathogenic mechanisms of Hypomyelinating Leukodystrophies (HLDs). Neurology International, 15(3), 1155-1173. [DOI:10.3390/neurolint15030072] [PMID]
Traverso, M., Assereto, S., Gazzerro, E., Savasta, S., Abdalla, E. M., & Rossi, A., et al. (2013). Novel FAM126A mutations in hypomyelination and congenital cataract disease. Biochemical and Biophysical Research Communications, 439(3), 369–372. [DOI:10.1016/j.bbrc.2013.08.077] [PMID]
Traverso, M., Yuregir, O. O., Mimouni-Bloch, A., Rossi, A., Aslan, H., & Gazzerro, E., et al. (2013). Hypomyelination and congenital cataract: identification of novel mutations in two unrelated families. European Journal of Paediatric Neurology : EJPN : Official Journal of the European Paediatric Neurology Society, 17(1), 108–111. [DOI:10.1016/j.ejpn.2012.06.004] [PMID]
Troncoso, M., Balut, F., Witting, S., Rubilar, C., Carrera, J., & Cartes, F., et al. (2021). Hypomyelination and Congenital Cataract: Identification of a Novel likely pathogenic c. 414+ 1G> A in FAM126A gene Variant. Clinical Case Reports, 9(5), e04171. [DOI:10.1002/ccr3.4171] [PMID]
Ugur, S. A., & Tolun, A. (2008). A deletion in DRCTNNB1A associated with hypomyelination and juvenile onset cataract. European Journal of Human Genetics, 16(2), 261-264. [DOI:10.1038/sj.ejhg.5201935] [PMID]
Ulengin-Talkish, I., Parson, M. A. H., Jenkins, M. L., Roy, J., Shih, A. Z. L., & St-Denis, N., et al. (2021). Palmitoylation targets the calcineurin phosphatase to the phosphatidylinositol 4-kinase complex at the plasma membrane. Nature Communications, 12(1), 6064. [DOI:10.1038/s41467-021-26326-4] [PMID]
Zara, F., Biancheri, R., Bruno, C., Bordo, L., Assereto, S., & Gazzerro, E., et al. (2006). Deficiency of hyccin, a newly identified membrane protein, causes hypomyelination and congenital cataract. Nature Genetics, 38(10), 1111–1113. [DOI:10.1038/ng1870] [PMID]
Zhang, Q., Zhang, B., Lim, N. K. H., Zhang, X., Meng, S., & Nyengaard, J. R., et al. (2022). Hyccin/FAM126A deficiency reduces glial enrichment and axonal sheath, which are rescued by overexpression of a plasma membrane-targeting PI4KIIIα in Drosophila. Biochemical and Biophysical Research Communications, 589, 71–77. [DOI:10.1016/j.bbrc.2021.11.106] [PMID]
Zhang, K., Lin, G., Han, D., Han, Y., Peng, R., & Li, J. (2022). Adaptation of ACMG-ClinGen Technical Standards for Copy Number Variant Interpretation Concordance. Frontiers in Genetics, 13, 829728. [DOI:10.3389/fgene.2022.829728] [PMID]
Type of Study: Original |
Subject:
Cellular and molecular Neuroscience
Received: 2024/09/10 | Accepted: 2025/01/20 | Published: 2025/03/1
Received: 2024/09/10 | Accepted: 2025/01/20 | Published: 2025/03/1
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
