

Volume 16, Issue 2 (March & April 2025)
BCN 2025, 16(2): 449-460 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Abrishami M, Rezaei-Tavirani M. Exploring the Role of MicroRNAs and Associated Proteins in Multiple Sclerosis. BCN 2025; 16 (2) :449-460
URL: http://bcn.iums.ac.ir/article-1-2864-en.html
URL: http://bcn.iums.ac.ir/article-1-2864-en.html



1- Department of Medical Laboratory Sciences, Student Research Committee, School of Allied Medical Sciences, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
2- Proteomics Research Center, School of Paramedical Sciences, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
2- Proteomics Research Center, School of Paramedical Sciences, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
Full-Text [PDF 1067 kb]
| Abstract (HTML)
Full-Text:
1. Introduction
Multiple sclerosis (MS) is a chronic autoimmune disease that affects the central nervous system (CNS). Due to complex pathogenesis and lack of specific biomarkers, diagnosis of MS is a challenge. Recent data show that the incidence of MS is increasing globally. Early diagnosis of MS reduces the burden of disability-adjusted life years and associated health care costs (Solomon et al., 2023).
Extensive exploration has demonstrated that MS is thought to be caused by systemic immune activation of autoimmune mechanisms against CNS components. In MS, inflammation is regulated by interactions between several immune cells, such as T and B cells, macrophages, and CNS glial cells (microglia and astrocytes), as well as antigens that react against myelin, especially myelin basic protein and myelin oligodendrocyte glycoprotein (Pietrasik et al., 2021). Additionally, Th1 and Th17 cells can cross the blood-brain barrier (BBB) and migrate to the CNS, then activate microglia and secretion of inflammatory cytokines, such as tumor necrosis factor-α (l), interleukin (IL)-1β and IL-6 (Murphy et al., 2010). Diagnosis of MS is based on clinical and radiological assessment, and current effective treatments target the peripheral immune system (Healy et al., 2022a; Luo et al., 2017; Inojosa et al., 2021). The diagnostic criteria for MS have limitations of sensitivity and specificity (Ian McDonald et al., 2001). If a specific test is lacking (Tavazzi et al., 2020), some patients may be misdiagnosed or not diagnosed (Ian McDonald et al., 2001; Tavazzi et al., 2020).
Noncoding RNAs (ncRNAs) are RNA molecules that do not encode proteins but play essential roles in gene expression regulation. Investigations indicate that ncRNAs are involved in the pathogenesis and progression of MS (Jalaiei et al., 2021; Zheleznyakova et al., 2021). The roles of ncRNAs in regulating immune cells and pathways, as well as their involvement in the neurodegeneration process and MS progression, are highlighted by researchers (Elkhodiry & El Tayebi 2021; Yousuf & Qurashi, 2021; Piket et al., 2019). In addition to their diagnostic potential, ncRNAs may serve as therapeutic targets for MS treatment. It may be effective in personalizing medicine (Gupta et al., 2019; Piket et al., 2019; Zheleznyakova et al., 2021; Chen et al., 2022). MicroRNAs (miRNAs) are small noncoding RNA molecules that regulate various cellular processes, including inflammation and immune response. They have emerged as potential biomarkers for MS, as they are stable, detectable, and quantifiable in various biological fluids, such as blood and cerebrospinal fluid. Several studies have shown that the expression levels of miRNAs are altered in MS patients compared to healthy controls or other neurological diseases. Moreover, some miRNAs have been associated with clinical features, such as disease duration, disability score, relapse rate, lesion load, inflammation, neurodegeneration, and treatment response (Gao et al., 2021; Yang et al., 2018; Slota et al., 2019; Yang et al., 2018; Gao et al., 2021; Minutti-Zanella., 2022).
The application of bioinformatics in the interpretation of genomics outcomes has attracted researchers’ attention. Network analysis is a bioinformatics tool suitable for interpreting genomics data (Ni et al., 2014). There are several documents about the application of network analysis in exploring MS’s molecular aspects (Hao et al., 2022). Protein-protein interaction (PPI) network analysis is a computational method to identify interactions between proteins and understand their functional relationships. In a published study, researchers used PPI network analysis to identify hub-long ncRNAs and potential drugs for MS. They constructed a PPI network using differentially expressed mRNAs and identified four modules enriched in immune-related pathways. They identified three key long ncRNAs (LINC00649, TP73-AS1, and MALAT1) associated with MS (Chakraborty et al., 2022; Gabrielli & Verderio, 2023; Khan et al., 2016; Yadav et al., 2023; Wang et al., 2022). The present study investigates the possible role of miRNAs and their related protein in diagnosing MS disorder via network analysis using data from the GEO database.
2. Materials and Methods
Data collection
MS microRNA data had been selected from the GEO database (GSE124900). Data were produced using the NGS approach. The 16 peripheral blood-extracted microRNA profiles from relapsing-remitting MS patients and 8 healthy controls (Baulina et al., 2019) were selected for more analysis. UMAP analysis revealed that the samples were not separated via the performed assessment. So, the suitable samples, including 4 patients and 4 controls, were candidates for evaluation. In addition to the microRNA analysis, related proteins have been investigated in the literature.
Pre-evaluation analysis
The GEO2R program was applied to evaluate data. Visualization tools such as Boxplot, Venn diagram, and volcano plot were employed to gain insights into potential microRNA expression signatures and identify differentially expressed microRNAs. These analytical tools are crucial in minimizing bias in microRNA expression levels and identifying potential diagnostic targets (microRNA) for future interventions. These visualization tools visually represent the data, enabling researchers to identify patterns and trends that may not be immediately apparent from raw data. The significant differentially expressed miRNAs with a P adj<0.05 were selected for further analysis.
PPI network analysis
Actions (activation, expression, catalysis, and post-translation) between the explored proteins were assessed via an action map using Cytoscape software, version 3.7.2. Furthermore, PPI network analysis has been employed to analyze the related proteins. The associated proteins were included in an interactome via the STRING database using Cytoscape software, version 3.7.2 (Shannon et al., 2003). The nodes were connected via undirected edges. The utilization of PPI network analysis has provided a comprehensive understanding of the binding relationship among proteins and has facilitated the identification of potential key proteins involved in diagnosing MS.
Integrating multiple analytical approaches, including miRNA profiling, related protein investigation, and PPI network analysis, has enabled a more comprehensive understanding of the molecular mechanisms underlying MS.
Statistical analysis
P adj<0.05 was applied to find the significant differentially expressed microRNAs. A confidence score of 0.4 was considered to form a PPI network.
3. Results
The extracted significant differentially expressed miRNAs are presented in Table 1.
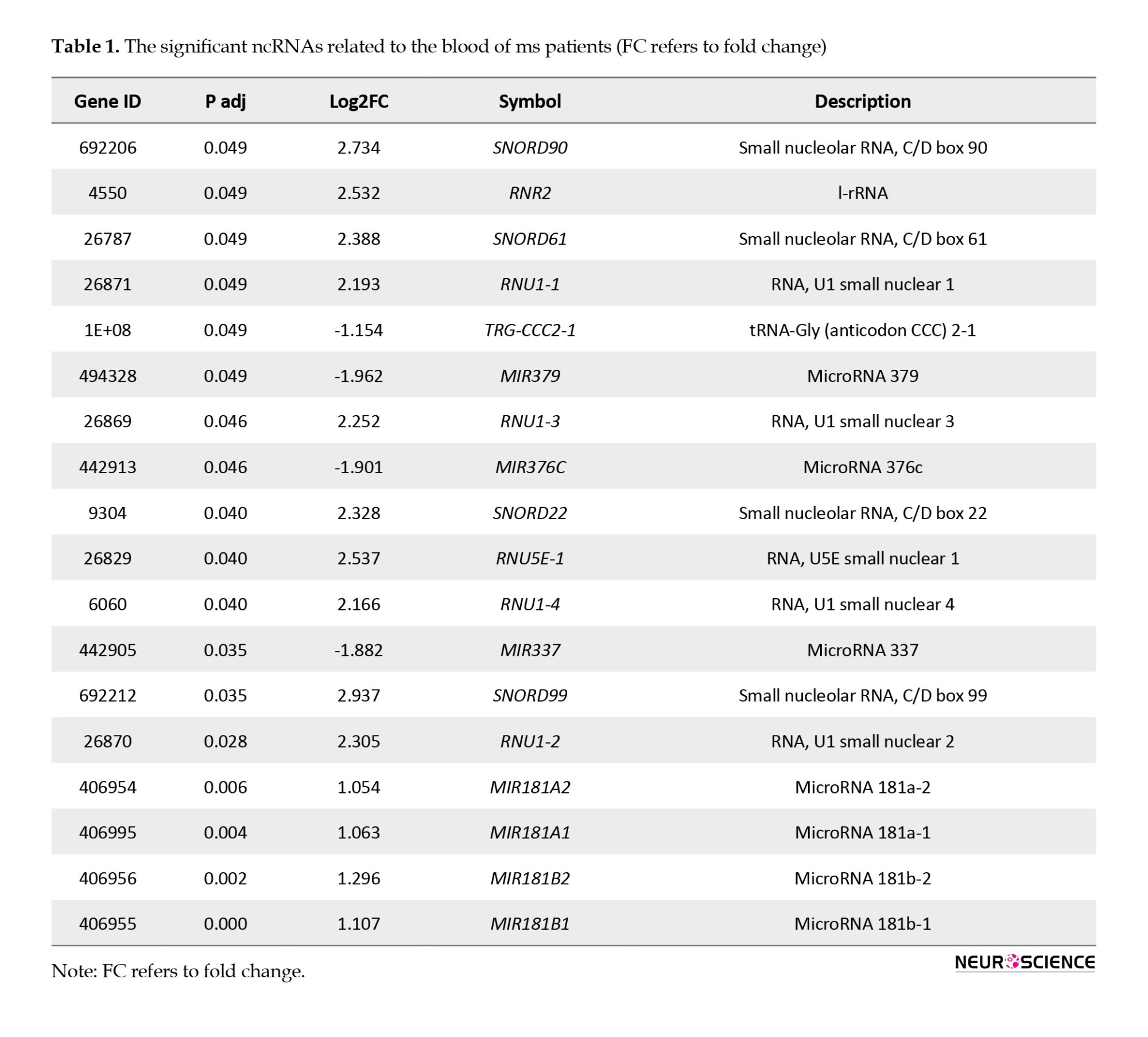
As shown in Table 1, 18 miRNAs are eligible for more investigation. Box plot (Figure 1) has been utilized to maintain that our selected miRNAs are trustworthy for further investigation because their median line was in the same position. The volcano plot was used to identify the significantly differentially expressed (DE) miRNAs between the two groups.
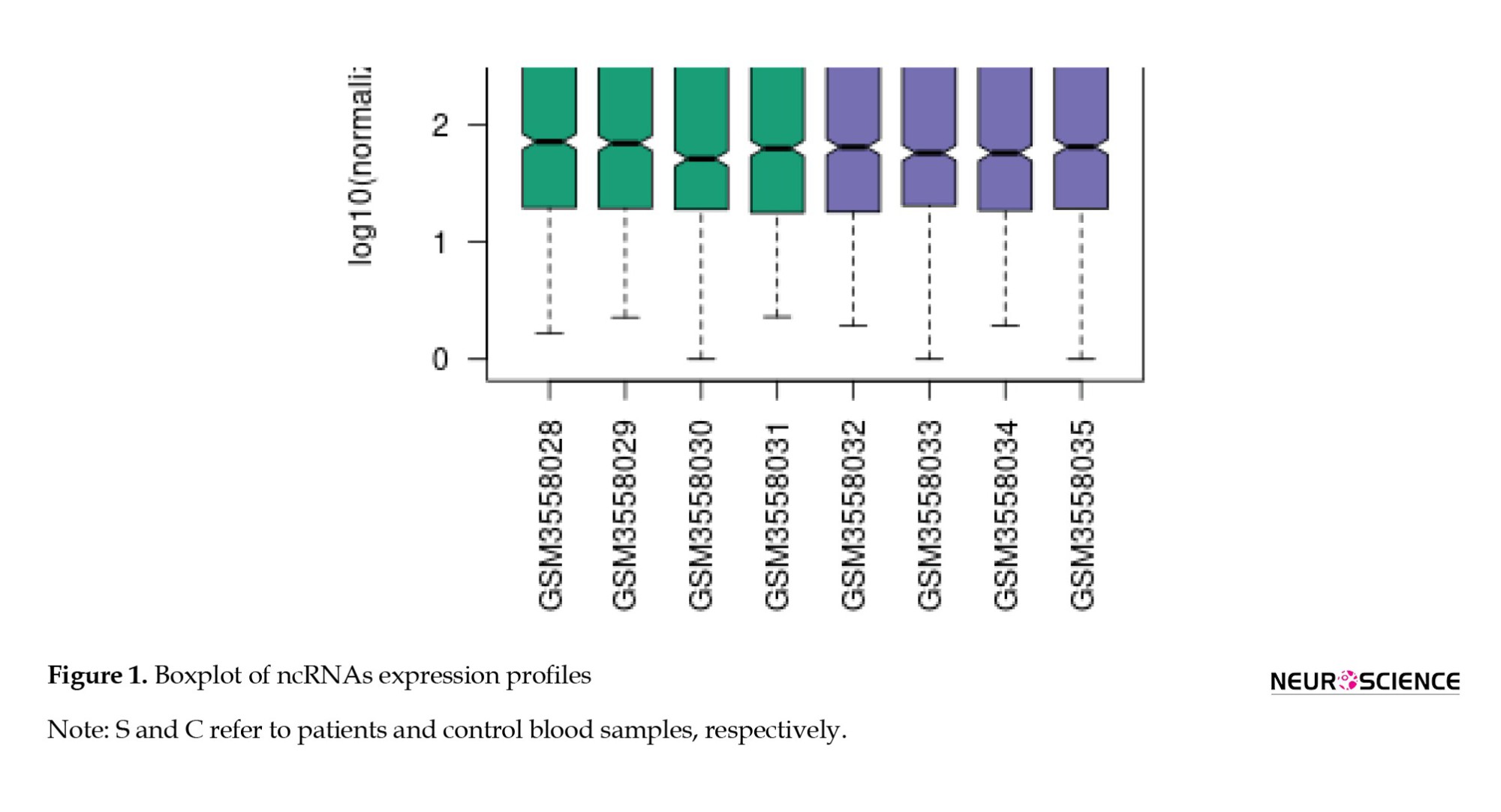
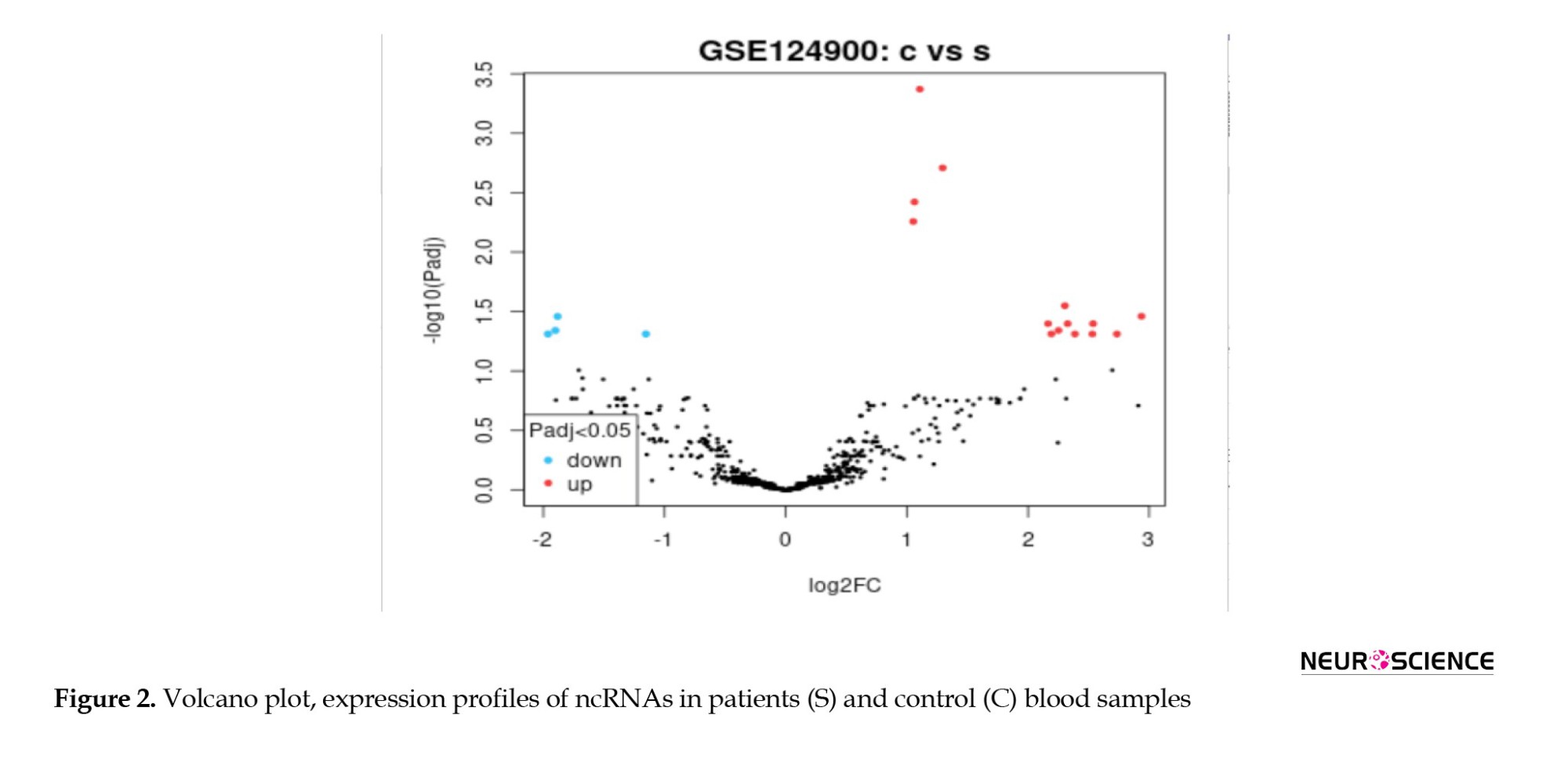
Figure 2 shows several significant DE miRNAs discriminate patient samples from controls.
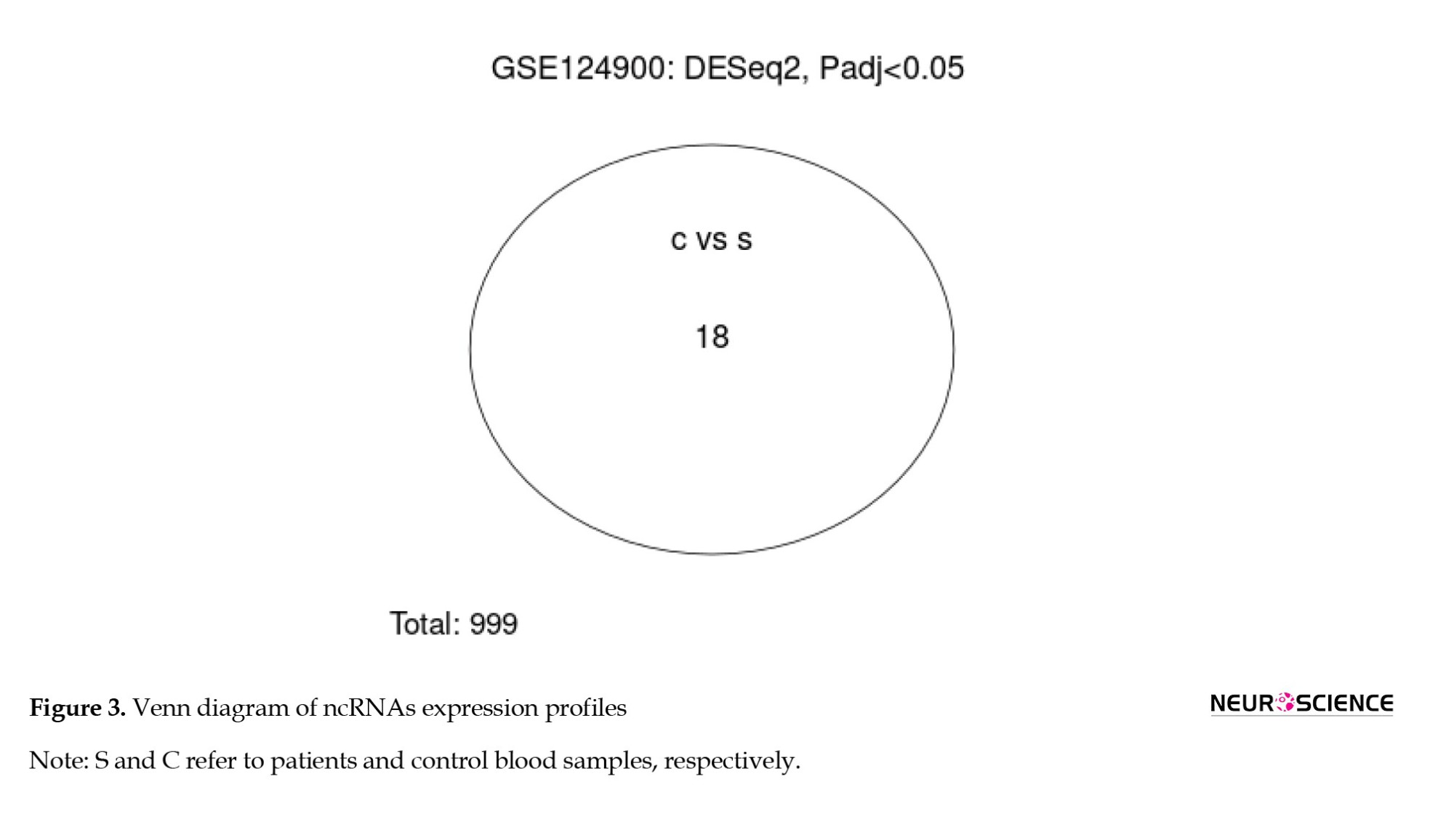
Venn diagram visualizes 18 significant DE miRNAs that differentiate the compare groups (Figure 3).
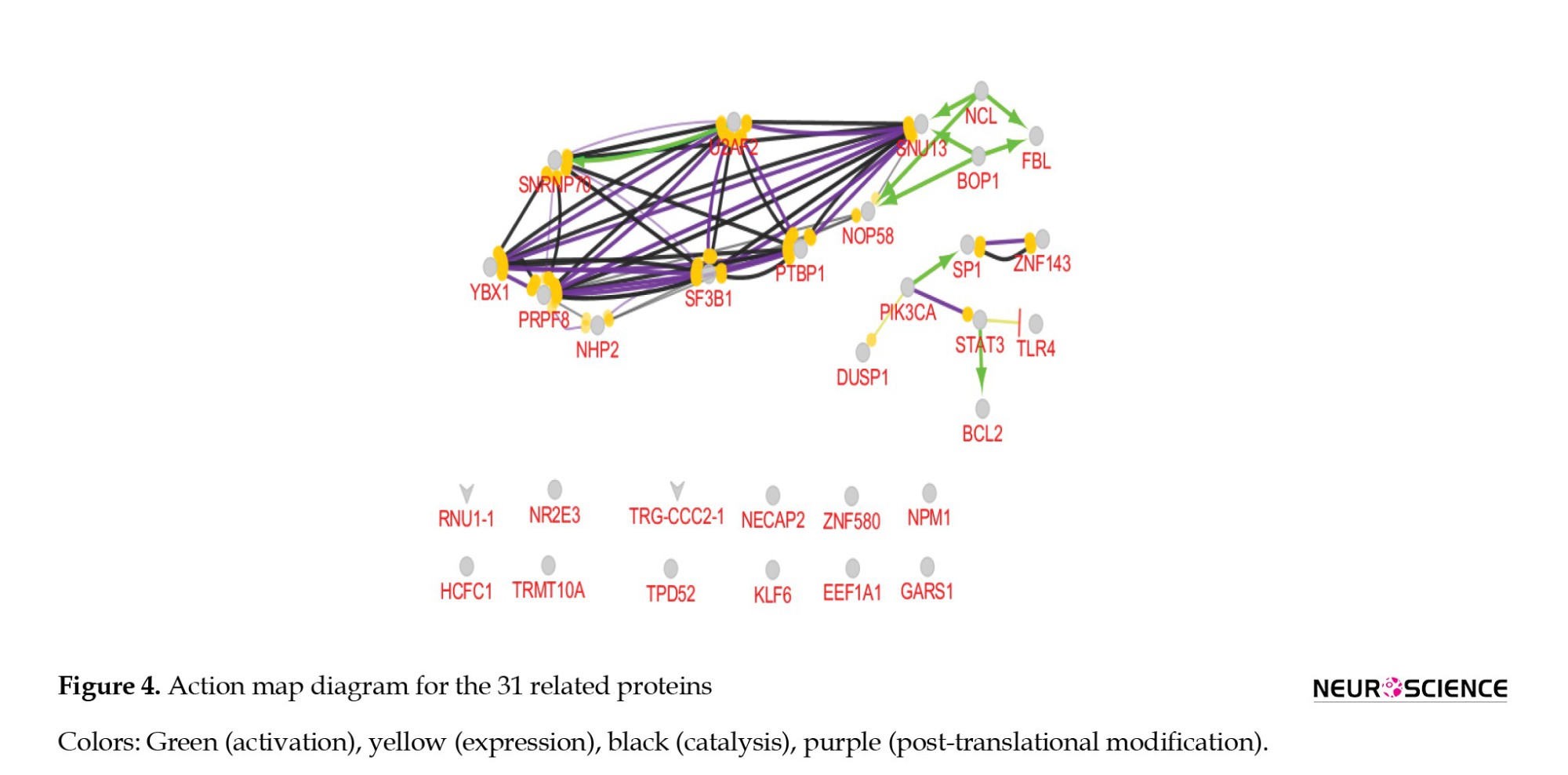
Furthermore, 31 proteins related to 18 microRNAs, including FBL, NCL, NHP2L1, PTBP1, YBX1, NOP58, PRPF8, SNRNP70, U2AF2, STAT3, SP1, SNRPA, TLR4, PIK3CA, BOP1, DUSP1, TPD52, ZNF580, NECAP2, ELF3, GARS, BCL2, TRMT10A, NR2E3, KLF3, ZNF143, HCFC1, EEFA1, SF3B1, NHP2, and NPM1 were identified from literature. An action map was constructed to illustrate the connections among the 31 related proteins. Each color in the diagram represents a different protein-protein function: Green, yellow, black, and purple, which consists of activation, expression, catalysis, and post-translation. It should be noted that functional interactions for 12 proteins have remained unknown. A thorough investigation determined that NPM1 is the most highly connected protein in this PPI diagram. NPM1 is connected with FBL, YBX1, NOP58, PRPF8, STAT3, BOP1, PIK3CA, and DUSP1 (Figure 4).
The PPI network, including 31 proteins, is represented in Figure 5. As is detected in Figure 5, 27 proteins are connected in the subnetwork, while the remaining individuals are isolated nodes. Nodes of the network are visualized based on degree value.

Considering the results of the action map and PPI network analysis, the carousal proteins were selected for more study. A list of the carousal proteins and their biological descriptions are presented in Table 2.

4. Discussion
MS is a complex autoimmune disease that affects the CNS. The exact mechanisms underlying MS pathogenesis are still elusive. miRNAs, RNA molecules that do not encode proteins but regulate gene expression, have been suggested to play a role in MS. Moreover, miRNAs are stable biomarkers in peripheral blood that make them appropriate candidates for diagnosing MS. They can also reflect the status of immune cells and inflammatory pathways that are relevant to MS pathogenesis, distinguish MS patients from healthy controls and other disease controls, as well as different MS subtypes and stages, and predict the response to treatment and the risk of relapse or progression in MS patients (Gandhi 2015; LoPresti 2022; Ziemssen et al., 2019). Therefore, by analyzing the related proteins of miRNAs, we can find critical pathways that can be the key to finding new treatments and diagnosing methods for MS.
The present study used data from the GEO database to identify miRNAs differentially expressed in MS patients compared to healthy controls. A total of 18 significantly dysregulated miRNAs were selected, and 31 related proteins involved in MS disease were determined for analysis. Pre-evaluation of data indicates that the analysis is valid. An action map was used as a tool to screen data. Based on action map findings, NCL, U2AF2, SNRNP70, YBX1, and STAT3 have considerable relationships with others. Activation of SNRNP70 by U2AF2 is an important event because it has a role in neuromuscular synapses, and U2AF2 also catalyzes and has an effect on post-translation of PTBP1 which has been investigated in literature as an MS biomarker (Masaki et al., 2020). STAT3 can be a potential candidate because of its relationship with IL-6, which was upregulated in MS patients (Razia et al., 2023).
SNRNP70 plays an important role in regulating motor axonal growth, nerve-dependent acetylcholine receptor clustering, neuromuscular synaptogenesis, and a protective role for a limited number of axonal transcripts, preventing them from degradation. Moreover, non-nuclear SNRNP70 can locally regulate splice variants of transcripts such as agrin, thereby locally controlling the formation of synapses (Lubelsky et al., 2021; Nakaya, 2020; Nikolaou et al., 2020).
On the other hand, YBX1 affects both PTBP1 and U2AF2 and also expresses PRPF8, which has a role in the post-transcription of U2AF2. It has a role in neuroinflammation signaling (Kloetgen et al., 2020). The relationship and effect of STAT3 (which has been investigated as an MS biomarker) and TLR4 are remarkable; they both have positive regulation of each other. It is essential to mention that TLR4 is a drug target because of its main role in inflammation and T cell and macrophage activities (Ahuja et al., 2020; Li et al., 2019; Mukherjee et al., 2019). TLR4 and BTK are interconnected in the context of immune signaling and inflammation, and both have been implicated in the pathogenesis of MS; TLR4 also has an impact on BTK, which is an MS drug target (Geladariset al., 2022; Krämer et al., 2023). The other protein, PIK3K, can be a potential biomarker as it has a relationship with mir-21 in activating the PI3K/AKT pathway as a possible therapy for treating MS (Manian et al., 2021; Yin et al., 2022). It is connected with STAT3 by its post-translation modification and expression.
PPI network analysis was performed to identify the most relevant proteins interacting with these microRNAs. Based on degree value, the results indicated that several proteins, including FBL, NCL, PTBP1, SNRNP70, YBX1, PRPF8, and NPM1, are important ones. Except for NPM1, the other vital nodes of the PPI network are highlighted in the action map. Other studies have also reported these proteins. For instance, PTBP1 has been shown to regulate alternative splicing of genes involved in immune response and inflammation (Babenko et al., 2022; Hensel et al., 2022), key pathways in MS pathogenesis (Hecker et al., 2019; Masaki et al., 2020). NPM1 has been implicated in regulating microglia activation, a hallmark of MS (Healy et al., 2022b; Jäntti et al., 2022; Sen et al., 2022).
PTB1 has been shown to regulate the expression of BDNF (Notaras & van den Buuse, 2019), which has been shown to promote neuronal growth and survival in the CNS, synaptic plasticity, and mitochondrial biogenesis, making it a promising biomarker in neurodegenerative conditions (Ahmed et al., 2021; Pisani et al., 2023).
SNRNP70 has been shown to regulate alternative splicing of genes involved in neuronal function and synaptic plasticity, which are disrupted in MS (Ksiazek-Winiarek et al., 2015; Nakaya, 2020; Nikolaou et al., 2020). YBX1 is identified as a common high central gene in PPI networks corresponding to both type 1 diabetes and MS (Safari-Alighiarloo et al., 2020). As discussed before, NOP58 is another candidate with a high degree and is also activated by BOP1 and NCL. NOP58 has a connection with PTBP1 and PRPF8.
5. Conclusion
Moreover, as PTBP1, STAT3, and TLR4 have been suggested in the literature as potential biomarkers for MS, based on our investigation with bioinformatics approaches, they have roles in regulating the expression and function of NCL, NOP58, SNRNP70, U2AF2, YBX1, PRPF8, BOP1, and PIK3K proteins. These proteins involve various cellular processes relevant to MS pathogenesis, such as RNA splicing, transcription, inflammation, and apoptosis. Therefore, we propose a new biomarker panel consisting of PTBP1, STAT3, and TLR4, as well as SNRNP70, YBX1, PTB1, and PI3K as candidate drug targets. In addition, we suggest that monitoring the effect of targeting TLR4 on STAT3 activity could be a useful strategy for developing novel therapeutics for MS. Further experimental and theoretical investigations are suggested to validate the proposed biomarker panel and drug targets. Limited sources of databases and samples are limitations of this study.
Ethical Considerations
Compliance with ethical guidelines
This study was approved by the Research Ethics Committee of Shahid Beheshti University of Medical Sciences, Tehran, Iran (Code: IR.SBMU.RETECH.REC.1402.727).
Funding
This study was funded by the Student Research Committee, Shahid Beheshti University of Medical Sciences, Tehran, Iran (Project No.: 1402-63026).
Authors' contributions
All authors contributed equally to the conception and design of the study, data collection and analysis, interception of the results and drafting of the manuscript. Each author approved the final version of the manuscript for submission.
Conflict of interest
The authors declared no conflict of interest.
Acknowledgments
The authors appreciate the “Student Research Committee” and “Research & Technology Chancellor” at Shahid Beheshti University of Medical Sciences, Tehran, Iran for their financial support of this study.
References
Multiple sclerosis (MS) is a chronic autoimmune disease that affects the central nervous system (CNS). Due to complex pathogenesis and lack of specific biomarkers, diagnosis of MS is a challenge. Recent data show that the incidence of MS is increasing globally. Early diagnosis of MS reduces the burden of disability-adjusted life years and associated health care costs (Solomon et al., 2023).
Extensive exploration has demonstrated that MS is thought to be caused by systemic immune activation of autoimmune mechanisms against CNS components. In MS, inflammation is regulated by interactions between several immune cells, such as T and B cells, macrophages, and CNS glial cells (microglia and astrocytes), as well as antigens that react against myelin, especially myelin basic protein and myelin oligodendrocyte glycoprotein (Pietrasik et al., 2021). Additionally, Th1 and Th17 cells can cross the blood-brain barrier (BBB) and migrate to the CNS, then activate microglia and secretion of inflammatory cytokines, such as tumor necrosis factor-α (l), interleukin (IL)-1β and IL-6 (Murphy et al., 2010). Diagnosis of MS is based on clinical and radiological assessment, and current effective treatments target the peripheral immune system (Healy et al., 2022a; Luo et al., 2017; Inojosa et al., 2021). The diagnostic criteria for MS have limitations of sensitivity and specificity (Ian McDonald et al., 2001). If a specific test is lacking (Tavazzi et al., 2020), some patients may be misdiagnosed or not diagnosed (Ian McDonald et al., 2001; Tavazzi et al., 2020).
Noncoding RNAs (ncRNAs) are RNA molecules that do not encode proteins but play essential roles in gene expression regulation. Investigations indicate that ncRNAs are involved in the pathogenesis and progression of MS (Jalaiei et al., 2021; Zheleznyakova et al., 2021). The roles of ncRNAs in regulating immune cells and pathways, as well as their involvement in the neurodegeneration process and MS progression, are highlighted by researchers (Elkhodiry & El Tayebi 2021; Yousuf & Qurashi, 2021; Piket et al., 2019). In addition to their diagnostic potential, ncRNAs may serve as therapeutic targets for MS treatment. It may be effective in personalizing medicine (Gupta et al., 2019; Piket et al., 2019; Zheleznyakova et al., 2021; Chen et al., 2022). MicroRNAs (miRNAs) are small noncoding RNA molecules that regulate various cellular processes, including inflammation and immune response. They have emerged as potential biomarkers for MS, as they are stable, detectable, and quantifiable in various biological fluids, such as blood and cerebrospinal fluid. Several studies have shown that the expression levels of miRNAs are altered in MS patients compared to healthy controls or other neurological diseases. Moreover, some miRNAs have been associated with clinical features, such as disease duration, disability score, relapse rate, lesion load, inflammation, neurodegeneration, and treatment response (Gao et al., 2021; Yang et al., 2018; Slota et al., 2019; Yang et al., 2018; Gao et al., 2021; Minutti-Zanella., 2022).
The application of bioinformatics in the interpretation of genomics outcomes has attracted researchers’ attention. Network analysis is a bioinformatics tool suitable for interpreting genomics data (Ni et al., 2014). There are several documents about the application of network analysis in exploring MS’s molecular aspects (Hao et al., 2022). Protein-protein interaction (PPI) network analysis is a computational method to identify interactions between proteins and understand their functional relationships. In a published study, researchers used PPI network analysis to identify hub-long ncRNAs and potential drugs for MS. They constructed a PPI network using differentially expressed mRNAs and identified four modules enriched in immune-related pathways. They identified three key long ncRNAs (LINC00649, TP73-AS1, and MALAT1) associated with MS (Chakraborty et al., 2022; Gabrielli & Verderio, 2023; Khan et al., 2016; Yadav et al., 2023; Wang et al., 2022). The present study investigates the possible role of miRNAs and their related protein in diagnosing MS disorder via network analysis using data from the GEO database.
2. Materials and Methods
Data collection
MS microRNA data had been selected from the GEO database (GSE124900). Data were produced using the NGS approach. The 16 peripheral blood-extracted microRNA profiles from relapsing-remitting MS patients and 8 healthy controls (Baulina et al., 2019) were selected for more analysis. UMAP analysis revealed that the samples were not separated via the performed assessment. So, the suitable samples, including 4 patients and 4 controls, were candidates for evaluation. In addition to the microRNA analysis, related proteins have been investigated in the literature.
Pre-evaluation analysis
The GEO2R program was applied to evaluate data. Visualization tools such as Boxplot, Venn diagram, and volcano plot were employed to gain insights into potential microRNA expression signatures and identify differentially expressed microRNAs. These analytical tools are crucial in minimizing bias in microRNA expression levels and identifying potential diagnostic targets (microRNA) for future interventions. These visualization tools visually represent the data, enabling researchers to identify patterns and trends that may not be immediately apparent from raw data. The significant differentially expressed miRNAs with a P adj<0.05 were selected for further analysis.
PPI network analysis
Actions (activation, expression, catalysis, and post-translation) between the explored proteins were assessed via an action map using Cytoscape software, version 3.7.2. Furthermore, PPI network analysis has been employed to analyze the related proteins. The associated proteins were included in an interactome via the STRING database using Cytoscape software, version 3.7.2 (Shannon et al., 2003). The nodes were connected via undirected edges. The utilization of PPI network analysis has provided a comprehensive understanding of the binding relationship among proteins and has facilitated the identification of potential key proteins involved in diagnosing MS.
Integrating multiple analytical approaches, including miRNA profiling, related protein investigation, and PPI network analysis, has enabled a more comprehensive understanding of the molecular mechanisms underlying MS.
Statistical analysis
P adj<0.05 was applied to find the significant differentially expressed microRNAs. A confidence score of 0.4 was considered to form a PPI network.
3. Results
The extracted significant differentially expressed miRNAs are presented in Table 1.
As shown in Table 1, 18 miRNAs are eligible for more investigation. Box plot (Figure 1) has been utilized to maintain that our selected miRNAs are trustworthy for further investigation because their median line was in the same position. The volcano plot was used to identify the significantly differentially expressed (DE) miRNAs between the two groups.
Figure 2 shows several significant DE miRNAs discriminate patient samples from controls.
Venn diagram visualizes 18 significant DE miRNAs that differentiate the compare groups (Figure 3).
Furthermore, 31 proteins related to 18 microRNAs, including FBL, NCL, NHP2L1, PTBP1, YBX1, NOP58, PRPF8, SNRNP70, U2AF2, STAT3, SP1, SNRPA, TLR4, PIK3CA, BOP1, DUSP1, TPD52, ZNF580, NECAP2, ELF3, GARS, BCL2, TRMT10A, NR2E3, KLF3, ZNF143, HCFC1, EEFA1, SF3B1, NHP2, and NPM1 were identified from literature. An action map was constructed to illustrate the connections among the 31 related proteins. Each color in the diagram represents a different protein-protein function: Green, yellow, black, and purple, which consists of activation, expression, catalysis, and post-translation. It should be noted that functional interactions for 12 proteins have remained unknown. A thorough investigation determined that NPM1 is the most highly connected protein in this PPI diagram. NPM1 is connected with FBL, YBX1, NOP58, PRPF8, STAT3, BOP1, PIK3CA, and DUSP1 (Figure 4).
The PPI network, including 31 proteins, is represented in Figure 5. As is detected in Figure 5, 27 proteins are connected in the subnetwork, while the remaining individuals are isolated nodes. Nodes of the network are visualized based on degree value.
Considering the results of the action map and PPI network analysis, the carousal proteins were selected for more study. A list of the carousal proteins and their biological descriptions are presented in Table 2.
4. Discussion
MS is a complex autoimmune disease that affects the CNS. The exact mechanisms underlying MS pathogenesis are still elusive. miRNAs, RNA molecules that do not encode proteins but regulate gene expression, have been suggested to play a role in MS. Moreover, miRNAs are stable biomarkers in peripheral blood that make them appropriate candidates for diagnosing MS. They can also reflect the status of immune cells and inflammatory pathways that are relevant to MS pathogenesis, distinguish MS patients from healthy controls and other disease controls, as well as different MS subtypes and stages, and predict the response to treatment and the risk of relapse or progression in MS patients (Gandhi 2015; LoPresti 2022; Ziemssen et al., 2019). Therefore, by analyzing the related proteins of miRNAs, we can find critical pathways that can be the key to finding new treatments and diagnosing methods for MS.
The present study used data from the GEO database to identify miRNAs differentially expressed in MS patients compared to healthy controls. A total of 18 significantly dysregulated miRNAs were selected, and 31 related proteins involved in MS disease were determined for analysis. Pre-evaluation of data indicates that the analysis is valid. An action map was used as a tool to screen data. Based on action map findings, NCL, U2AF2, SNRNP70, YBX1, and STAT3 have considerable relationships with others. Activation of SNRNP70 by U2AF2 is an important event because it has a role in neuromuscular synapses, and U2AF2 also catalyzes and has an effect on post-translation of PTBP1 which has been investigated in literature as an MS biomarker (Masaki et al., 2020). STAT3 can be a potential candidate because of its relationship with IL-6, which was upregulated in MS patients (Razia et al., 2023).
SNRNP70 plays an important role in regulating motor axonal growth, nerve-dependent acetylcholine receptor clustering, neuromuscular synaptogenesis, and a protective role for a limited number of axonal transcripts, preventing them from degradation. Moreover, non-nuclear SNRNP70 can locally regulate splice variants of transcripts such as agrin, thereby locally controlling the formation of synapses (Lubelsky et al., 2021; Nakaya, 2020; Nikolaou et al., 2020).
On the other hand, YBX1 affects both PTBP1 and U2AF2 and also expresses PRPF8, which has a role in the post-transcription of U2AF2. It has a role in neuroinflammation signaling (Kloetgen et al., 2020). The relationship and effect of STAT3 (which has been investigated as an MS biomarker) and TLR4 are remarkable; they both have positive regulation of each other. It is essential to mention that TLR4 is a drug target because of its main role in inflammation and T cell and macrophage activities (Ahuja et al., 2020; Li et al., 2019; Mukherjee et al., 2019). TLR4 and BTK are interconnected in the context of immune signaling and inflammation, and both have been implicated in the pathogenesis of MS; TLR4 also has an impact on BTK, which is an MS drug target (Geladariset al., 2022; Krämer et al., 2023). The other protein, PIK3K, can be a potential biomarker as it has a relationship with mir-21 in activating the PI3K/AKT pathway as a possible therapy for treating MS (Manian et al., 2021; Yin et al., 2022). It is connected with STAT3 by its post-translation modification and expression.
PPI network analysis was performed to identify the most relevant proteins interacting with these microRNAs. Based on degree value, the results indicated that several proteins, including FBL, NCL, PTBP1, SNRNP70, YBX1, PRPF8, and NPM1, are important ones. Except for NPM1, the other vital nodes of the PPI network are highlighted in the action map. Other studies have also reported these proteins. For instance, PTBP1 has been shown to regulate alternative splicing of genes involved in immune response and inflammation (Babenko et al., 2022; Hensel et al., 2022), key pathways in MS pathogenesis (Hecker et al., 2019; Masaki et al., 2020). NPM1 has been implicated in regulating microglia activation, a hallmark of MS (Healy et al., 2022b; Jäntti et al., 2022; Sen et al., 2022).
PTB1 has been shown to regulate the expression of BDNF (Notaras & van den Buuse, 2019), which has been shown to promote neuronal growth and survival in the CNS, synaptic plasticity, and mitochondrial biogenesis, making it a promising biomarker in neurodegenerative conditions (Ahmed et al., 2021; Pisani et al., 2023).
SNRNP70 has been shown to regulate alternative splicing of genes involved in neuronal function and synaptic plasticity, which are disrupted in MS (Ksiazek-Winiarek et al., 2015; Nakaya, 2020; Nikolaou et al., 2020). YBX1 is identified as a common high central gene in PPI networks corresponding to both type 1 diabetes and MS (Safari-Alighiarloo et al., 2020). As discussed before, NOP58 is another candidate with a high degree and is also activated by BOP1 and NCL. NOP58 has a connection with PTBP1 and PRPF8.
5. Conclusion
Moreover, as PTBP1, STAT3, and TLR4 have been suggested in the literature as potential biomarkers for MS, based on our investigation with bioinformatics approaches, they have roles in regulating the expression and function of NCL, NOP58, SNRNP70, U2AF2, YBX1, PRPF8, BOP1, and PIK3K proteins. These proteins involve various cellular processes relevant to MS pathogenesis, such as RNA splicing, transcription, inflammation, and apoptosis. Therefore, we propose a new biomarker panel consisting of PTBP1, STAT3, and TLR4, as well as SNRNP70, YBX1, PTB1, and PI3K as candidate drug targets. In addition, we suggest that monitoring the effect of targeting TLR4 on STAT3 activity could be a useful strategy for developing novel therapeutics for MS. Further experimental and theoretical investigations are suggested to validate the proposed biomarker panel and drug targets. Limited sources of databases and samples are limitations of this study.
Ethical Considerations
Compliance with ethical guidelines
This study was approved by the Research Ethics Committee of Shahid Beheshti University of Medical Sciences, Tehran, Iran (Code: IR.SBMU.RETECH.REC.1402.727).
Funding
This study was funded by the Student Research Committee, Shahid Beheshti University of Medical Sciences, Tehran, Iran (Project No.: 1402-63026).
Authors' contributions
All authors contributed equally to the conception and design of the study, data collection and analysis, interception of the results and drafting of the manuscript. Each author approved the final version of the manuscript for submission.
Conflict of interest
The authors declared no conflict of interest.
Acknowledgments
The authors appreciate the “Student Research Committee” and “Research & Technology Chancellor” at Shahid Beheshti University of Medical Sciences, Tehran, Iran for their financial support of this study.
References
Ahmed, S., Kwatra, M., Gawali, B., Panda, S. R., & Naidu, V. G. M. (2021). Potential role of TrkB agonist in neuronal survival by promoting CREB/BDNF and PI3K/Akt signaling in vitro and in vivo model of 3-nitropropionic acid (3-NP)-induced neuronal death. Apoptosis : An International Journal on Programmed Cell Death, 26(1-2), 52–70. [DOI:10.1007/s10495-020-01645-x] [PMID]
Ahuja, A., Kim, E., Sung, G. H., & Cho, J. Y. (2020). STAT3 Differentially regulates TLR4-mediated inflammatory responses in early or late phases. International Journal of Molecular Sciences, 21(20), 7675. [DOI:10.3390/ijms21207675] [PMID]
Alhazzani, K., Ahmad, S. F., Al-Harbi, N. O., Attia, S. M., Bakheet, S. A., & Sarawi, W., et al. (2021). Pharmacological Inhibition of STAT3 by Stattic Ameliorates Clinical Symptoms and Reduces Autoinflammation in Myeloid, Lymphoid, and Neuronal Tissue Compartments in Relapsing-Remitting Model of Experimental Autoimmune Encephalomyelitis in SJL/J Mice. Pharmaceutics, 13(7), 925. [PMID]
Appu, A. P., Bagh, M. B., Sadhukhan, T., Mondal, A., Casey, S., & Mukherjee, A. B. (2019). Cln3-mutations underlying juvenile neuronal ceroid lipofuscinosis cause significantly reduced levels of Palmitoyl-protein thioesterases-1 (Ppt1)-protein and Ppt1-enzyme activity in the lysosome. Journal of Inherited Metabolic Disease, 42(5), 944–954. [DOI:10.1002/jimd.12106] [PMID]
Babenko, V. N., Shishkina, G. T., Lanshakov, D. A., Sukhareva, E. V., & Dygalo, N. N. (2022). LPS administration impacts glial immune programs by alternative splicing. Biomolecules, 12(2), 277. [DOI:10.3390/biom12020277] [PMID]
Banskota, S., & Adamson, D. C. (2021). Pituitary Adenomas: From diagnosis to therapeutics. Biomedicines, 9(5), 494. [DOI:10.3390/biomedicines9050494] [PMID]
Baulina, N., Osmak, G., Kiselev, I., Popova, E., Boyko, A., & Kulakova, O., et al. (2019). MiRNAs from DLK1-DIO3 Imprinted Locus at 14q32 are associated with multiple sclerosis: Gender-specific expression and regulation of receptor tyrosine kinases signaling. Cells, 8(2), 133. [DOI:10.3390/cells8020133] [PMID]
Chakraborty, S., Tarasankar, M., Sushmita, B., & Soumili, S. (2022). Identifying common genes, proteins, and pathways from human mirna and gene blood profiles in multiple sclerosis patients. bioRxiv. [DOI:10.1101/2022.11.29.518394]
Chen, B., Dragomir, M. P., Yang, C., Li, Q., Horst, D., & Calin, G. A. (2022). Targeting non-coding RNAs to overcome cancer therapy resistance. Signal Transduction and Targeted Therapy, 7(1), 121. [PMID]
Cvitkovic, I., & Jurica, M. S. (2013). Spliceosome database: a tool for tracking components of the spliceosome. Nucleic Acids Research, 41(Database issue), D132–D141. [DOI:10.1093/nar/gks999] [PMID]
Elkhodiry, A. A., & El Tayebi, H. M. (2021). Scavenging the hidden impacts of non-coding RNAs in multiple sclerosis. Non-Coding RNA Research, 6(4), 187–199. [DOI:10.1016/j.ncrna.2021.12.002] [PMID]
Falini, B., Brunetti, L., Sportoletti, P., & Martelli, M. P. (2020). NPM1-mutated acute myeloid leukemia: From bench to bedside. Blood, 136(15), 1707–1721. [DOI:10.1182/blood.2019004226] [PMID]
Folch, J., Busquets, O., Ettcheto, M., Sánchez-López, E., Castro-Torres, R. D., & Verdaguer, E., et al. (2018). Memantine for the Treatment of Dementia: A review on its current and future applications. Journal of Alzheimer's disease : JAD, 62(3), 1223–1240. [PMID]
Frankish, A., Carbonell-Sala, S., Diekhans, M., Jungreis, I., Loveland, J. E., & Mudge, J. M., et al. (2023). GENCODE: Reference annotation for the human and mouse genomes in 2023. Nucleic Acids Research, 51(D1), D942–D949. [DOI:10.1093/nar/gkac1071] [PMID]
Fruman, D. A., Chiu, H., Hopkins, B. D., Bagrodia, S., Cantley, L. C., & Abraham, R. T. (2017). The PI3K pathway in human disease. Cell, 170(4), 605–635. [DOI:10.1016/j.cell.2017.07.029] [PMID]
Gabrielli, M., & Verderio, C. (2023). Exosomal profiling should be used to monitor disease activity in MS patients: Commentary. Multiple Sclerosis (Houndmills, Basingstoke, England), 29(10), 1208. [DOI:10.1177/13524585231195858] [PMID]
Gandhi, R. (2015). miRNA in multiple sclerosis: Search for novel biomarkers. Multiple Sclerosis (Houndmills, Basingstoke, England), 21(9), 1095–1103. [DOI:10.1177/1352458515578771] [PMID]
Gao, Y., Han, D., & Feng, J. (2021). MicroRNA in multiple sclerosis. Clinica Chimica Acta; International Journal of Clinical Chemistry, 516, 92–99. [DOI:10.1016/j.cca.2021.01.020] [PMID]
Geladaris, A., Torke, S., & Weber, M. S. (2022). Bruton's tyrosine kinase inhibitors in multiple sclerosis: Pioneering the path towards treatment of progression? CNS Drugs, 36(10), 1019–1030. [DOI:10.1007/s40263-022-00951-z] [PMID]
Gupta, M., Martens, K., Metz, L. M., de Koning, A. J., & Pfeffer, G. (2019). Long noncoding RNAs associated with phenotypic severity in multiple sclerosis. Multiple Sclerosis and Related Disorders, 36, 101407. [DOI:10.1016/j.msard.2019.101407] [PMID]
Hao, Y., He, M., Fu, Y., Zhao, C., Xiong, S., & Xu, X. (2022). Identification of novel key genes and pathways in multiple sclerosis based on weighted gene coexpression network analysis and long noncoding RNA-Associated competing endogenous RNA network. Oxidative Medicine and Cellular Longevity, 2022, 9328160. [DOI:10.1155/2022/9328160] [PMID]
Healy, L. M., Stratton, J. A., Kuhlmann, T., & Antel, J. (2022). The role of glial cells in multiple sclerosis disease progression. Nature reviews. Neurology, 18(4), 237–248. [DOI:10.1038/s41582-022-00624-x] [PMID]
Healy, L. M., Stratton, J. A., Kuhlmann, T., & Antel, J. (2022). The role of glial cells in multiple sclerosis disease progression. Nature reviews. Neurology, 18(4), 237–248. [DOI:10.1038/s41582-022-00624-x] [PMID]
Hecker, M., Rüge, A., Putscher, E., Boxberger, N., Rommer, P. S., & Fitzner, B., et al. (2019). Aberrant expression of alternative splicing variants in multiple sclerosis - A systematic review. Autoimmunity Reviews, 18(7), 721–732. [DOI:10.1016/j.autrev.2019.05.010] [PMID]
Hensel, J. A., Nicholas, S. E., Kimble, A. L., Nagpal, A. S., Omar, O. M. F., & Tyburski, J. D., et al. (2022). Splice factor polypyrimidine tract-binding protein 1 (Ptbp1) primes endothelial inflammation in atherogenic disturbed flow conditions. Proceedings of the National Academy of Sciences of the United States of America, 119(30), e2122227119. [DOI:10.1073/pnas.2122227119] [PMID]
Inojosa, H., Proschmann, U., Akgün, K., & Ziemssen, T. (2021). A focus on secondary progressive multiple sclerosis (SPMS): Challenges in diagnosis and definition. Journal of Neurology, 268(4), 1210–1221. [DOI:10.1007/s00415-019-09489-5] [PMID]”
Manoochehrabadi, S., Arsang-Jang, S., Mazdeh, M., Inoko, H., Sayad, A., & Taheri, M. (2019). Analysis of STAT1, STAT2 and STAT3 mRNA expression levels in the blood of patients with multiple sclerosis. Human Antibodies, 27(2), 91–98. [PMID]
McDonald, W. I., Compston, A., Edan, G., Goodkin, D., Hartung, H. P., & Lublin, F. D., et al. (2001). Recommended diagnostic criteria for multiple sclerosis: Guidelines from the International Panel on the diagnosis of multiple sclerosis. Annals of Neurology, 50(1), 121–127. [DOI:10.1002/ana.1032] [PMID]
National Institute of Allergy and Infectious Diseases. STAT3 Gain-of-Function Disease. Maryland: National Institute of Allergy and Infectious Diseases; 2016. [Link]
Jalaiei, A., Asadi, M. R., Sabaie, H., Dehghani, H., Gharesouran, J., & Hussen, B. M., et al. (2021). Long non-coding RNAs, Novel offenders or guardians in multiple sclerosis: A scoping review. Frontiers in Immunology, 12, 774002. [DOI:10.3389/fimmu.2021.774002] [PMID]
Jäntti, H., Sitnikova, V., Ishchenko, Y., Shakirzyanova, A., Giudice, L., & Ugidos, I. F., et al. (2022). Microglial amyloid beta clearance is driven by PIEZO1 channels. Journal of Neuroinflammation, 19(1), 147. [DOI:10.1186/s12974-022-02486-y] [PMID]
Kasapkara, Ç. S., Ceylan, A. C., Yılmaz, D., Kıreker Köylü, O., Yürek, B., & Civelek Ürey, B., et al. (2023). CLN3-Associated NCL case with a preliminary diagnosis of niemann pick type C. Molecular Syndromology, 14(1), 30–34. [DOI:10.1159/000525100] [PMID]
Khan, N., Mironov, G., & Berezovski, M. V. (2016). Direct detection of endogenous MicroRNAs and their post-transcriptional modifications in cancer serum by capillary electrophoresis-mass spectrometry. Analytical and Bioanalytical Chemistry, 408(11), 2891–2899. [DOI:10.1007/s00216-015-9277-y] [PMID]
Kim, H. J., Kim, H., Lee, J. H., & Hwangbo, C. (2023). Toll-like receptor 4 (TLR4): New insight immune and aging. Immunity & Ageing : I & A, 20(1), 67. [DOI:10.1186/s12979-023-00383-3] [PMID]
Kloetgen, A., Duggimpudi, S., Schuschel, K., Hezaveh, K., & Picard, D., Schaal, H., et al. (2020). YBX1 Indirectly Targets Heterochromatin-Repressed Inflammatory Response-Related Apoptosis Genes through Regulating CBX5 mRNA. International Journal of Molecular Sciences, 21(12), 4453. [PMID]
Kong, E., Sucic, S., Monje, F. J., Reisinger, S. N., Savalli, G., & Diao, W., et al. (2015). Corrigendum: STAT3 controls IL6-dependent regulation of serotonin transporter function and depression-like behaviour. Scientific Reports, 5, 11965. [DOI:10.1038/srep11965] [PMID]
Krämer, J., Bar-Or, A., Turner, T. J., & Wiendl, H. (2023). Bruton tyrosine kinase inhibitors for multiple sclerosis. Nature Reviews. Neurology, 19(5), 289–304. [DOI:10.1038/s41582-023-00800-7] [PMID]
Ksiazek-Winiarek, D. J., Szpakowski, P., & Glabinski, A. (2015). Neural plasticity in multiple sclerosis: The functional and molecular background. Neural Plasticity, 2015, 307175. [DOI:10.1155/2015/307175] [PMID]
Li, H., Liu, S., Han, J., Li, S., Gao, X., & Wang, M., et al. (2021). Role of toll-like receptors in neuroimmune diseases: Therapeutic targets and problems. Frontiers in Immunology, 12, 777606. [DOI:10.3389/fimmu.2021.777606] [PMID]
Li, Z. H., Wang, Y. F., He, D. D., Zhang, X. M., Zhou, Y. L., & Yue, H., et al. (2019). Let-7f-5p suppresses Th17 differentiation via targeting STAT3 in multiple sclerosis. Aging, 11(13), 4463–4477. [DOI:10.18632/aging.102093] [PMID]
Loh, J. T., Lee, K. G., Lee, A. P., Teo, J. K. H., Lim, H. L., & Kim, S. S., et al. (2021). DOK3 maintains intestinal homeostasis by suppressing JAK2/STAT3 signaling and S100a8/9 production in neutrophils. Cell Death & Disease, 12(11), 1054. [PMID]
LoPresti, P. (2022). Serum-Based biomarkers in neurodegeneration and multiple sclerosis. Biomedicines, 10(5), 1077.[DOI:10.3390/biomedicines10051077] [PMID]
Lubelsky, Y., Zuckerman, B., & Ulitsky, I. (2021). High-resolution mapping of function and protein binding in an RNA nuclear enrichment sequence. The EMBO Journal, 40(12), e106357. [DOI:10.15252/embj.2020106357] [PMID]
Luo, C., Jian, C., Liao, Y., Huang, Q., Wu, Y., & Liu, X., et al. (2017). The role of microglia in multiple sclerosis. Neuropsychiatric Disease and Treatment, 13, 1661–1667. [DOI:10.2147/NDT.S140634] [PMID]
Manian, M., Sohrabi, E., Eskandari, N., Assarehzadegan, M. A., Ferns, G. A., & Nourbakhsh, M., et al. (2021). An integrated bioinformatics analysis of the potential regulatory effects of miR-21 on T-cell related target genes in multiple sclerosis. Avicenna Journal of Medical Biotechnology, 13(3), 149–165.[DOI:10.18502/ajmb.v13i3.6364] [PMID]
Masaki, K., Sonobe, Y., Ghadge, G., Pytel, P., Lépine, P., & Pernin, F., et al. (2020). RNA-binding protein altered expression and mislocalization in MS. Neurology(R) Neuroimmunology & Neuroinflammation, 7(3), e704. [DOI:10.1212/NXI.0000000000000704] [PMID]
Miranda-Hernandez, S., & Baxter, A. G. (2013). Role of toll-like receptors in multiple sclerosis. American Journal of Clinical and Experimental Immunology, 2(1), 75–93. [PMID]
Minutti-Zanella, C., Bojalil-Álvarez, L., García-Villaseñor, E., López-Martínez, B., Pérez-Turrent, M., & Murrieta-Álvarez, I., et al. (2022). miRNAs in multiple sclerosis: A clinical approach. Multiple Sclerosis and Related Disorders, 63, 103835. [DOI:10.1016/j.msard.2022.103835] [PMID]
Mukherjee, S., Huda, S., & Sinha Babu, S. P. (2019). Toll-like receptor polymorphism in host immune response to infectious diseases: A review. Scandinavian Journal of Immunology, 90(1), e12771. [DOI:10.1111/sji.12771] [PMID]
Murphy, A. C., Lalor, S. J., Lynch, M. A., & Mills, K. H. (2010). Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain, Behavior, and Immunity, 24(4), 641–651. [DOI:10.1016/j.bbi.2010.01.014] [PMID]
Nakaya, T. (2020). Dissection of FUS domains involved in regulation of SnRNP70 gene expression. FEBS Letters, 594(21), 3518–3529. [DOI:10.1002/1873-3468.13924] [PMID]
Nikolaou, N., Gordon, P. M., Hamid, F., Taylor, R., Makeyev, E. V., & Houart, C. (2020). Cytoplasmic pool of spliceosome protein SNRNP70 regulates the axonal transcriptome and development of motor connectivity. [Preprint].[DOI:10.1101/2020.05.25.097444]
Ni, Y., Stingo, F. C., & Baladandayuthapani, V. (2014). Integrative bayesian network analysis of genomic data. Cancer Informatics, 13(Suppl 2), 39–48. [DOI:10.4137/CIN.S13786] [PMID]
Notaras, M., & van den Buuse, M. (2019). Brain-Derived Neurotrophic Factor (BDNF): Novel insights into regulation and genetic variation. The Neuroscientist : A Review Journal Bringing Neurobiology, Neurology and Psychiatry, 25(5), 434–454. [DOI:10.1177/1073858418810142] [PMID]
Pietrasik, S., Dziedzic, A., Miller, E., Starosta, M., & Saluk-Bijak, J. (2021). Circulating miRNAs as potential biomarkers distinguishing relapsing-remitting from secondary progressive multiple sclerosis. A review. International Journal of Molecular Sciences, 22(21), 11887. [DOI:10.3390/ijms222111887] [PMID]
Piket, E., Zheleznyakova, G. Y., Kular, L., & Jagodic, M. (2019). Small non-coding RNAs as important players, biomarkers and therapeutic targets in multiple sclerosis: A comprehensive overview. Journal of Autoimmunity, 101, 17–25. [DOI:10.1016/j.jaut.2019.04.002] [PMID]
Pisani, A., Paciello, F., Del Vecchio, V., Malesci, R., De Corso, E., & Cantone, E., et al. (2023). The Role of BDNF as a biomarker in cognitive and sensory neurodegeneration. Journal of Personalized Medicine, 13(4), 652. [DOI:10.3390/jpm13040652] [PMID]
Prabhu, L., Hartley, A. V., Martin, M., Warsame, F., Sun, E., & Lu, T. (2015). Role of post-translational modification of the Y box binding protein 1 in human cancers. Genes & Diseases, 2(3), 240–246. [DOI:10.1016/j.gendis.2015.05.001] [PMID]
Razia, R., Majeed, F., Amin, R., Mukhtar, S., Mahmood, K., & Abualait, T., et al. (2023). Predictive value of α-synuclein expression in peripheral blood of multiple sclerosis patients: A two-dimensional assessment of a selected biomarker. Plos One, 18(8), e0285022. [DOI:10.1371/journal.pone.0285022] [PMID]
Safari-Alighiarloo, N., Taghizadeh, M., Mohammad Tabatabaei, S., Namaki, S., & Rezaei-Tavirani, M. (2020). Identification of common key genes and pathways between type 1 diabetes and multiple sclerosis using transcriptome and interactome analysis. Endocrine, 68(1), 81–92. [DOI:10.1007/s12020-019-02181-8] [PMID]
Schott, G., Galarza-Muñoz, G., Trevino, N., Chen, X., & Weirauch, M., et al. (2021). U2AF2 binds IL7R exon 6 ectopically and represses its inclusion. RNA (New York, N.Y.), 27(5), 571–583. [PMID]
Sen, M. K., Mahns, D. A., Coorssen, J. R., & Shortland, P. J. (2022). The roles of microglia and astrocytes in phagocytosis and myelination: Insights from the cuprizone model of multiple sclerosis. Glia, 70(7), 1215–1250. [DOI:10.1002/glia.24148] [PMID]
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., & Ramage, D., et al. (2003). Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Research, 13(11), 2498–2504. [DOI:10.1101/gr.1239303] [PMID]
Shi, Y., Ding, Y., Li, G., Wang, L., Osman, R. A., & Sun, J., et al. (2021). Discovery of novel biomarkers for diagnosing and predicting the progression of multiple sclerosis using TMT-Based Quantitative Proteomics. Frontiers in Immunology, 12, 700031. [DOI:10.3389/fimmu.2021.700031] [PMID]
Shubina, M. Y., Arifulin, E. A., Sorokin, D. V., Sosina, M. A., Tikhomirova, M. A., & Serebryakova, M. V., et al. (2020). The GAR domain integrates functions that are necessary for the proper localization of fibrillarin (FBL) inside eukaryotic cells. PeerJ, 8, e9029. [DOI:10.7717/peerj.9029] [PMID]
Solomon, A. J., Marrie, R. A., Viswanathan, S., Correale, J., Magyari, M., & Robertson, N. P., et al. (2023). Global barriers to the diagnosis of multiple sclerosis: Data from the Multiple Sclerosis International Federation Atlas of MS, Third Edition. Neurology, 101(6), e624–e635. [DOI:10.1212/WNL.0000000000207481] [PMID]
Slota, J. A., & Booth, S. A. (2019). MicroRNAs in Neuroinflammation: Implications in Disease Pathogenesis, Biomarker Discovery and Therapeutic Applications. Non-coding RNA, 5(2), 35. [PMID]
Sun, G., Wang, Z., Ti, Y., Wang, Y., & Wang, J., Zhao, J., et al. (2017). STAT3 promotes bone fracture healing by enhancing the FOXP3 expression and the suppressive function of regulatory T cells. APMIS : Acta Pathologica, Microbiologica, et Immunologica Scandinavica, 125(8), 752–760. [DOI:10.1111/apm.12706] [PMID]
Tavazzi, E., Zivadinov, R., Dwyer, M. G., Jakimovski, D., Singhal, T., & Weinstock-Guttman, B., et al. (2020). MRI biomarkers of disease progression and conversion to secondary-progressive multiple sclerosis. Expert Review of Neurotherapeutics, 20(8), 821–834. [DOI:10.1080/14737175.2020.1757435] [PMID]
Thol, F., Kade, S., Schlarmann, C., Löffeld, P., Morgan, M., & Krauter, J., et al. (2012). Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood, 119(15), 3578–3584.[DOI:10.1182/blood-2011-12-399337] [PMID]
Vanhaesebroeck, B., Guillermet-Guibert, J., Graupera, M., & Bilanges, B. (2010). The emerging mechanisms of isoform-specific PI3K signalling. Nature reviews. Molecular Cell Biology, 11(5), 329–341. [DOI:10.1038/nrm2882] [PMID]
Vedi, M., Smith, J. R., Thomas Hayman, G., Tutaj, M., Brodie, K. C., & De Pons, J. L., et al. (2023). 2022 updates to the Rat Genome Database: a Findable, Accessible, Interoperable, and Reusable (FAIR) resource. Genetics, 224(1), iyad042. [DOI:10.1093/genetics/iyad042] [PMID]
Vogel, T. P., Leiding, J. W., Cooper, M. A., & Forbes Satter, L. R. (2023). STAT3 gain-of-function syndrome. Frontiers in Pediatrics, 10, 770077. [PMID]
Wang, T., Xu, S., Liu, L., Li, S., Zhang, H., Lu, X., et al. (2022). Integrated analysis of differentially expressed genes and a ceRNA network to identify hub lncRNAs and potential drugs for multiple sclerosis. American Journal of Translational Research, 14(2), 772–787. [PMID]
Xiang, M. (2013). Physiological and pharmacological regulation of the STAT3 pathway in cancer. [PhD dissertation]. Massachusetts: Harvard University. [Link]
Yadav, S. K., Jauhari, A., Singh, N., Pandey, A., Sarkar, S., & Pandey, S., et al. (2023). Transcriptomics and proteomics approach for the identification of altered blood microRNAs and plasma proteins in parkinson's disease. Cellular and Molecular Neurobiology, 43(7), 3527–3553. [DOI:10.1007/s10571-023-01362-4] [PMID]
Yang, X., Wu, Y., Zhang, B., & Ni, B. (2018). Noncoding RNAs in multiple sclerosis. Clinical Epigenetics, 10(1), 149. [DOI:10.1186/s13148-018-0586-9] [PMID]
Yin, X., Rang, X., Hong, X., Zhou, Y., Xu, C., & Fu, J. (2022). Immune cells transcriptome-based drug repositioning for multiple sclerosis. Frontiers in Immunology, 13, 1020721.[DOI:10.3389/fimmu.2022.1020721] [PMID]
Yousuf, A., & Qurashi, A. (2021). Non-coding RNAs in the Pathogenesis of Multiple Sclerosis. Frontiers in Genetics, 12, 717922. [PMID]
Zeinalzadeh, E., Valerievich Yumashev, A., Rahman, H. S., Marofi, F., Shomali, N., & Kafil, H. S., et al. (2021). The role of Janus Kinase/STAT3 pathway in hematologic malignancies with an emphasis on epigenetics. Frontiers in Genetics, 12, 703883. [DOI:10.3389/fgene.2021.703883] [PMID]
Zheng, C., Chen, J., Chu, F., Zhu, J., & Jin, T. (2020). Inflammatory Role of TLR-MyD88 Signaling in Multiple Sclerosis. Frontiers in Molecular Neuroscience, 12, 314. [DOI:10.3389/fnmol.2019.00314] [PMID]
Zheleznyakova, G. Y., Piket, E., Needhamsen, M., Hagemann-Jensen, M., Ekman, D., & Han, Y., et al. (2021). Small noncoding RNA profiling across cellular and biofluid compartments and their implications for multiple sclerosis immunopathology. Proceedings of the National Academy of Sciences of the United States of America, 118(17), e2011574118. [DOI:10.1073/pnas.2011574118] [PMID]
Zhou, X., Yuan, B., Tian, Y., Zhou, J., Wang, M., & Shakir, I., et al. (2023). Wiskott-Aldrich Syndrome protein regulates nucleolar organization and function in innate immune response. BioRxiv. [DOI:10.1101/2023.08.15.553387]
Ziemssen, T., Akgün, K., & Brück, W. (2019). Molecular biomarkers in multiple sclerosis. Journal of Neuroinflammation, 16(1), 272. [DOI:10.1186/s12974-019-1674-2] [PMID]
Zuurbier, C. J., Jong, W. M., Eerbeek, O., Koeman, A., Pulskens, W. P., & Butter, L. M., et al. (2012). Deletion of the innate immune NLRP3 receptor abolishes cardiac ischemic preconditioning and is associated with decreased Il-6/STAT3 signaling. Plos One, 7(7), e40643. [DOI:10.1371/journal.pone.0040643] [PMID]
Type of Study: Original |
Subject:
Behavioral Neuroscience
Received: 2024/01/16 | Accepted: 2024/04/8 | Published: 2025/03/1
Received: 2024/01/16 | Accepted: 2024/04/8 | Published: 2025/03/1
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
