

Volume 15, Issue 6 (November & December 2024)
BCN 2024, 15(6): 865-877 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Sakhaei A, Mohammadi-Asl J. Pathogenic CAPN3 Variant Identified in an Iranian Family With Limb-girdle Muscular Dystrophy. BCN 2024; 15 (6) :865-877
URL: http://bcn.iums.ac.ir/article-1-2833-en.html
URL: http://bcn.iums.ac.ir/article-1-2833-en.html



1- Department of Genetics, Faculty of Biological Sciences, North Tehran Branch, Islamic Azad University, Tehran, Iran.
2- Cancer, Petroleum and Environmental Pollutants Research Center, Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran.
2- Cancer, Petroleum and Environmental Pollutants Research Center, Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran.
Keywords: Limb-girdle muscular dystrophy (LGMDs) type 2A, Whole-exome sequencing (WES), Calpain-3, Missense variant
Full-Text [PDF 3447 kb]
| Abstract (HTML)
Full-Text:
1. Introduction
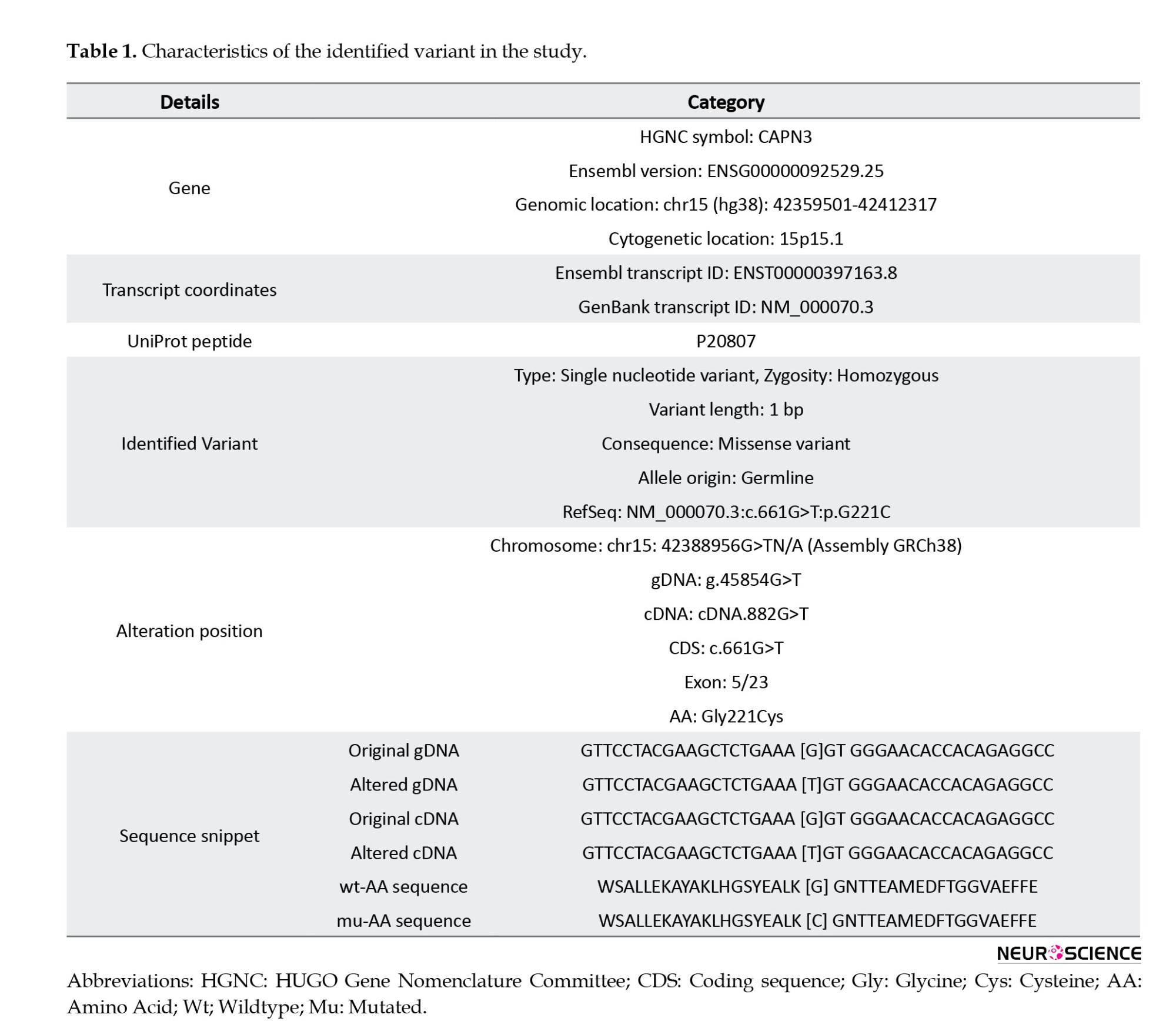
Clinically and genetically, limb-girdle muscular dystrophies (LGMDs) are a diverse group of muscle abnormalities that can be passed down in an autosomal recessive or dominant pattern (Bockhorst & Wicklund, 2020). On clinical examination, patients have symmetrical weakness in the pelvic, scapular, and trunk muscles. According to evidence, over 30 different genetic subtypes of LGMD have been discovered (Straub et al., 2018). Autosomal recessive limb-girdle muscular dystrophy-1 (LGMDR1; MIM:253600; ORPHA:267), also referred to as limb-girdle muscular dystrophy type 2A (LGMD2A), is the most common form of LGMD characterized by the progressive weakness of the proximal limb muscles and elevated serum creatine kinase (CK) levels (Nascimbeni et al., 2010). The evidence indicates that LGMDR1 is the first human disease discovered to be caused by mutations in the calcium-activated neutral proteinase 3 (CAPN3) gene (Chro. 15q15.1, OMIM: 114240). The gene encodes the calpain-3 protein, accounting for about 30% of all LGMD cases (Martinez-Thompson et al., 2018; Sorimachi et al., 2010). Although the facial and neck muscles are unaffected, the clinical features of LGMD2A are highly heterogeneous, present at any age (from infancy to adulthood), and include running difficulties, a waddling gait, scapular winging, as well as respiratory failure in the advanced stages of the disease (Hunter et al., 2019; Pozsgai et al., 2021). LGMD cases involving cardiac muscles have also been reported sporadically (Mori-Yoshimura et al., 2017; Narayanaswami et al., 2014). The CAPN3 gene product (non-lysosomal Ca2+-dependent cysteine protease), which consists of four domains (protease core subdomain-1, protease core subdomain-2, C2 domain-like domain, and penta-EF-hand domain) and 821 amino acids, cleaves and breaks down numerous essential skeletal myoproteins, particularly those banded with the structure of myofibrils (Ono et al., 2016) (Figure 1). This breakdown is vital for muscle maintenance and regeneration, as it allows for removing damaged or old proteins and replacing them with new ones. Calpain-3 also forms and matures muscle fibers during development and regulates muscle contraction. Physiological aberrations caused by CAPN3 defects can result in insufficient ryanodine regulation, leading to movement anomalies and muscular weakness (Taghizadeh et al., 2019).

Based on the Human Gene Mutation Database (HGMD) and Leiden Open Variation Database (LOVD), more than 500 different CAPN3 variants have been identified in LGMD cases. Investigations indicate that missense variants are the most typical, comprising more than half of all CAPN3 mutations and reported as mainly compound heterozygous (Fanin et al., 2014; Park et al., 2016). Each variant affects different amino acids and can cause diverse consequences in the calpain-3 protein sequence, disrupting its function. Many patients may be misdiagnosed in the early stages due to the high clinical and genetic heterogeneity of LGMDs. Effective genetic diagnosis is required for proper genetic counseling and treatment of LGMD patients (Taghizadeh et al., 2019). Because of the wide range of LGMD-causing variants, next-generation sequencing (NGS) is one of the best options for the conclusive differential diagnosis of LGMD (Fanin et al., 2009; Piluso et al., 2005). However, the causal mutation for more than half of LGMD cases remains unknown despite recent advancements in molecular diagnostic techniques (Angelini, 2020). In the current study, whole-exome sequencing (WES) was utilized to detect LGMD-causing variants in the proband, followed by Sanger sequencing to confirm and segregate the detected variation in the proband and the family members. The WES results revealed a pathogenic homozygous variant within the CAPN3 gene in the proband, which was confirmed and co-segregated with LGMD2A in the family. This study is the first report of this variant in LGMD cases, enriching our knowledge concerning LGMD pathology and facilitating the future clinical management of these patients.
2. Materials and Methods
Electroneurographic, laboratory, and clinical findings
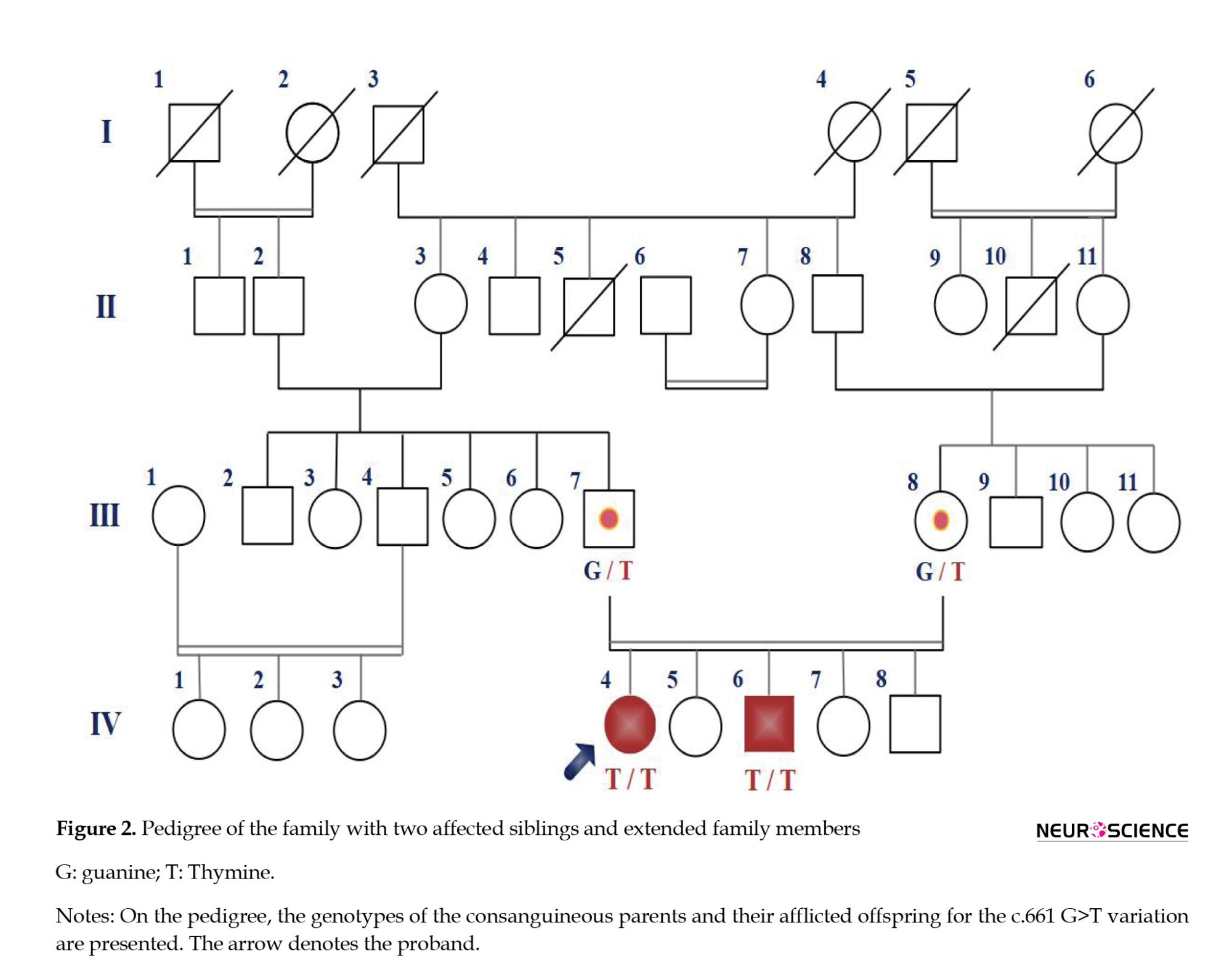
A consanguineous couple with two affected offspring strongly suspected of LGMD was referred to our Genetic Department at Ahvaz Noorgene Genetic and Clinical Laboratory to diagnose their muscular dystrophy type. The parents were first cousins, with two affected and three healthy offspring (Figure 2). The proband, a 33-year-old woman, was the oldest affected member in this family with a history of progressive muscular weakness that started at age 13. Her younger brother, a 27-year-old man, was another affected member whose progressive muscle weakness appeared since 16. Both affected had transient growing pains, sit-to-stand difficulties, a waddling gait, and mild proximal muscle weakness without involving facial muscles. Neither had intellectual disabilities, but calf pseudohypertrophy was present only in the proband. Though neither parent showed any symptoms of muscular weakness, the father was undergoing treatment for bladder cancer during the study. Additionally, the proband was getting therapy for an ovarian cyst. LGMD was suggested to be the first-line diagnosis by clinicians based on clinical presentations, electromyography (EMG), and laboratory tests.
The proband’s evoked EMG and nerve conduction velocity (NCV) examinations at the age of 13 demonstrated short-duration motor units, early recruitment, and wasting of the deltoids bilaterally with 2+ fibrillations. Electroneurography revealed the significant weakness of deltoids (grade 2/5) and positive sharp waves at rest with a brief duration (3-6 ms, NCV: 33 m/s). Laboratory parameters, including serum potassium, erythrocyte sedimentation rate, and C-reactive protein level, were all within normal ranges. However, the creatinine phosphokinase level was significantly elevated at 1187 U/L (reference range: 24-170 U/L). We applied WES to investigate the proband’s exome and identify the causal mutations in this LGMD2A-suspected family.
WES
After providing the proband and his family with an explanation of the NGS procedure, informed consent was obtained from them. Genomic DNA was isolated from the whole blood of the proband and all family members using a salting-out standard protocol in the presence of ethylenediaminetetraacetic acid (EDTA) as an anticoagulant. A NanoDrop200 spectrophotometer was used to determine the gDNA’s optical density (Thermo Fisher Scientific, Inc.). Exon capture and enrichment were conducted on genomic DNA using the Agilent SureSelectXT Human All Exon V8 (Agilent, Santa Clara, CA), and whole coding areas were sequenced with an average depth of 100x on an Illumina HiSeq 2000 sequencer (Illumina, Inc., San Diego, CA, USA) at the Noorgene Genetic and Clinical Laboratory. The raw data of WES was evaluated using a Core i7 central processing unit and the Linux (Ubuntu 20.04) operating system. As detailed below, various in silico prediction methods and bioinformatics resources were employed to provide additional support for the identified variants:
Quality control of the read lengths was evaluated by the FastQC program (version 0.11.9) based on a GC content of 50% and a Phred score of 20. Trimmomatic software (version 0.36) was used to eliminate adapter-contaminated and low-quality readings.
The Burrows–Wheeler Aligner (BWA) aligning tool (version 2.4.0) was used to precisely map sequence reads to the human reference genome (GRCh38/hg19), and outputs were generated in sequence alignment/map (SAM) file format. Afterward, the SAM file was converted to a binary alignment map file using the Picard program.
The local realignment of insertion/deletions (indels), base quality score recalibration, single-nucleotide variations (SNVs)/indels calling, and data compression were accomplished using a combination of genome analysis tool HaplotypeCaller and VarScan mpileup2snp protocol.
Gene ontology reference genome project, multiple association network integration method, protein analysis through evolutionary relationships, and Ensembl variant effect predictor (VEP) were used to annotate the output VCF file (Ensembl VEP).
Using the R programming command-line program, the annotated variations were filtered in the following ways.
Databases dbSNP v137, 1000 genomes project, Exome Sequencing Project, Human Gene Mutation Database (HGMD), Genome Aggregation Database (gnomAD), Iranome, and the Exome Aggregation Consortium (ExAC) were used to annotate the minor allele frequencies of all known variations.
Utilizing CADD (combined annotation-dependent depletion), PHRE, and GERP (the genomic evolutionary rate profiling) scores, we could filter out synonymous, deeply intronic, or less conserved variations.
Using the Phenolyzer (phenotype-based prioritization) tool and the Human Phenotype Ontology (HPO), we could separate the pathogenic variants from the non-pathogenic ones based on the presented clinical/paraclinical results.
According to standards established by the American College of Medical Genetics and Genomics Association for Molecular Pathology (ACMG/AMP), the remaining variations were analyzed and categorized. Besides, pathogenicity prediction of varieties was carried out through different bioinformatic tools: PolyPhen-2, SIFT, Pathogenic Mutation Prediction (PMut), and the MutationTaster.
Finally, the Integrative Genomics Viewer (IGV) program scrutinized the depth of reads for each variation.
Confirmation of variant and family segregation
The variation discovered through WES was confirmed by Sanger sequencing using an ABI3130xl sequencer and a BigDye Terminator v.3.1 Cycle Sequencing Kit (Life Technologies, USA), which also allowed for the investigation of family segregation. Primer3 web-based software was used to design the primer sequences spanning the WES-identified variation in the proband. Both the forward-primer (CAPN3-EX5-F278: 5′-ATT GTT TCC ATC CCA TGA GC-3′) and the reverse-primer (CAPN3-EX5-R278: 5′-TGA GAA ATT CCC AGT CCT CAA-3′) were put through the OligoAnalyzer tool to investigate their properties and evaluate their quality. Following the PCR amplification for the area containing the WES-identified variation, Sanger sequencing was conducted to confirm the potential causal variant in the proband and her family members.
The targeted area was amplified by running a PCR reaction in a total volume of 25 μL comprising 250 nM dNTPs, 100 ng of template DNA, 0.5 mM of each primer, and 1.25 units AmpliTaq Gold DNA polymerase in 1×reaction buffer on a FlexCycler Thermocycler (10 mM Tris HCl, pH 8.3, 50 mM KCl, 2.5 mM MgCl2). PCR procedure was conducted as follows: Initial denaturation at 95 ⁰C for 3 minutes, followed by 35 cycles of denaturation at 95 ⁰C for 45 seconds, annealing at 58 ⁰C for 1 minute, and extension at 72 ⁰C for 45 seconds, followed by a final 5-minute extension at 72 ⁰C. Purified PCR products were sequenced using a BigDye Terminator v.3.1 Cycle Sequencing Kit with inclined primers and PCR conditions. CodonCode aligner software was utilized to assemble and align Sanger sequencing files (version 9.0.1).
3. Results
WES data and co-segregation analysis
Variants within genes known to be involved in muscular dystrophies were prioritized for WES data analysis in this study. WES data were filtered to exclude non-genetic variants and compared with the databases. Following the WES data analysis of known muscular dystrophy genes with a recessive inheritance pattern and in silico analysis, we discovered a pathogenic homozygous variant within the CAPN3 gene, NM_000070.3:exon 5:c.661>GT, that had not previously been reported to the LGMD phenotypes in public databases (Table 1).
As shown in Figure 3, glycine at position 221 is highly conserved across species, emphasizing its critical role in protein function. The substitution of glycine with cysteine in this position disrupts the protein’s structure and function.

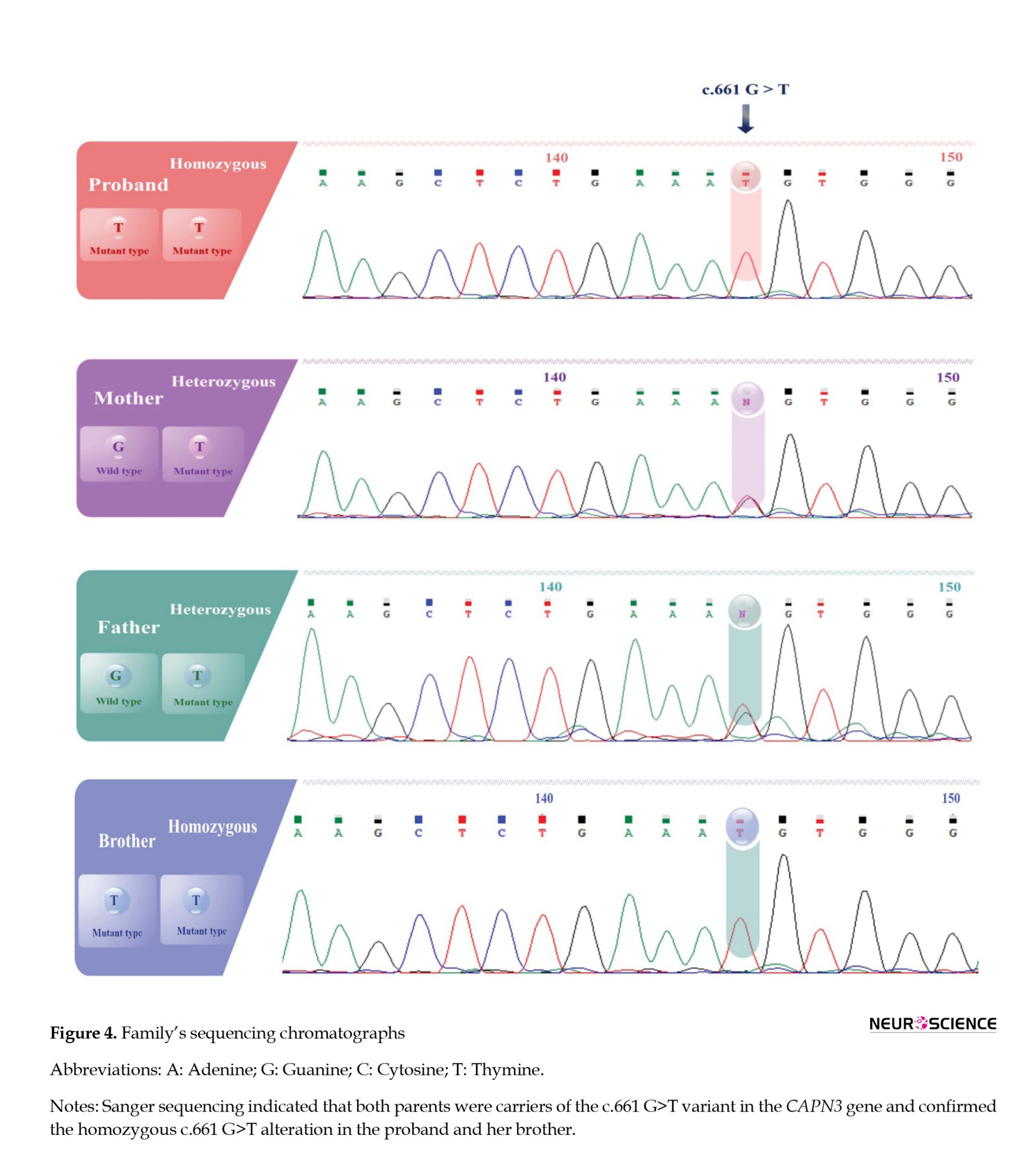
Subsequently, the pathogenic CAPN3 variant was validated through Sanger sequencing and segregated with LGMDA2 in the proband’s family (Figure 4). The WES and Sanger sequencing results consistently revealed that a GGT>TGT substitution at c.661 in exon 5 of the proband’s coding sequence (CDS) exists as a single nucleotide variant (SNV), and he was homozygous for this variant (TGT/TGT). Co-segregation analysis indicated that the proband’s brother was also homozygous for this variation (TGT/TGT), and both afflicted offspring had heterozygous carrier parents (GGT/TGT). In contrast, the healthy siblings did not carry the variant and were homozygous for the wild type (G) in the NM_000070.3:c.661 (GGT/GGT). This variation has not been reported as associated with LGMD in databases such as the HGMD, ESP, ExAC browser, LOVD, and dbSNP. In conclusion, the CAPN3 variation was segregated with LGMDA2 in this family, suggesting it could be considered the proband disease-causing.
Variant analysis
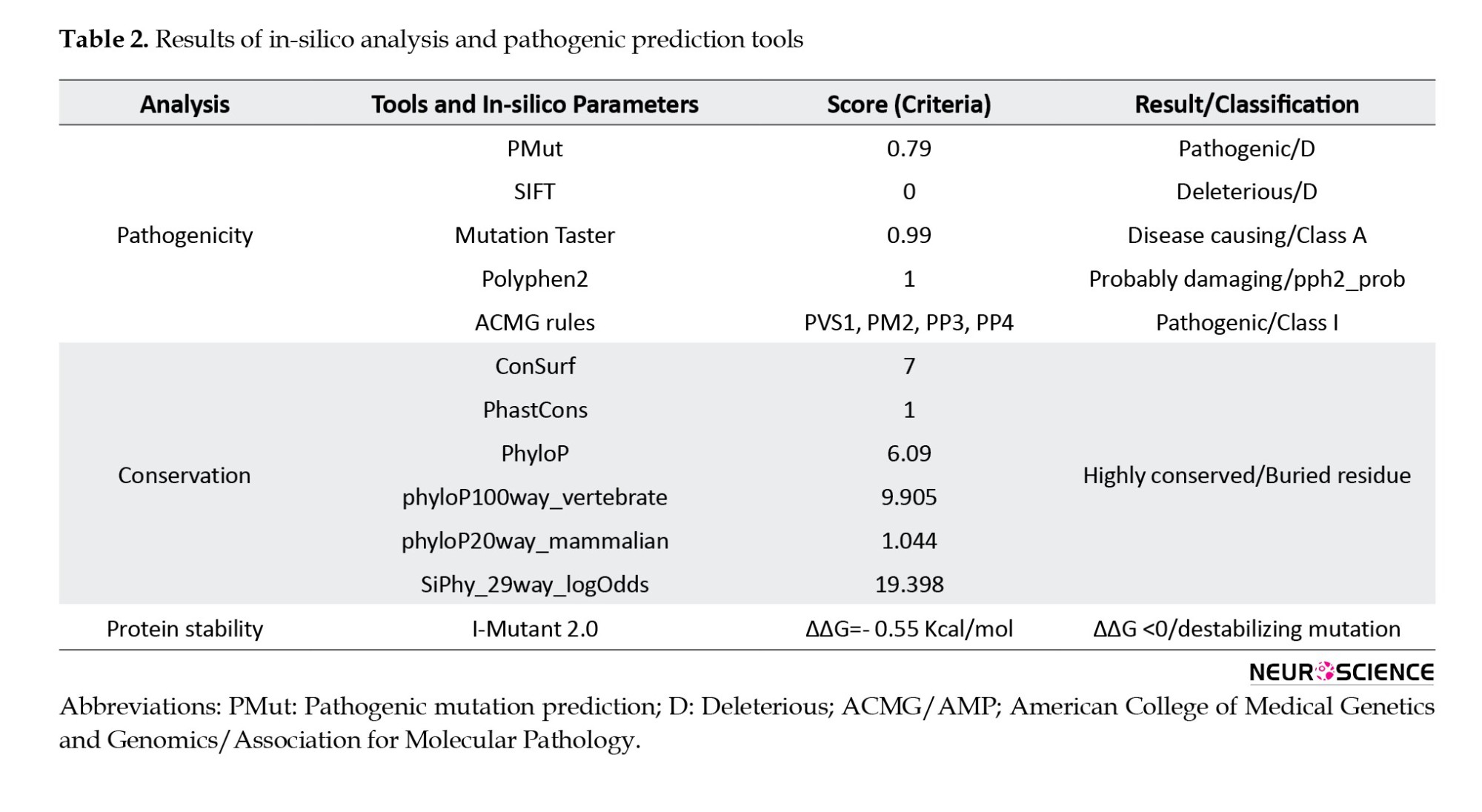
The missense variant c.661G>T:p.Gly221Cys resulted in a single nucleotide alternation (G>T) at site 661 in the coding region of CAPN3 (NM_000070.3), which introduced a glycine (Gly: [G]) residue that replaced a cysteine (Cys: [C]) at position 221 of the protein sequence (Table 1). Multiple sequence alignment analysis of the CAPN3 protein revealed that p.Gly221 is a highly conserved amino acid across many species (tfbsConsSites-score:940, phyloP100way_vertebrate: 9.905, phyloP20way_mammalian: 1.044, SiPhy_29way_logOdds: 19.398) (Table 2). The detected variant is topologically located in the peptidase_C2 domain, a highly conserved region, emphasizing the importance of the changed amino acid (glycine to cysteine) in protein function (Figure 3). The I-Mutant2.0 server, which computes the ΔΔG values of variations (DG “new protein”- DG “wildtype”), determined that the p.Gly221Cys substitution reduces the stability of the CAPN3 protein (ΔΔG=-0.55 kcal/mol, ΔΔG <0: Decrease stability). The ConSurf service also ran a conservation analysis on G.221, reporting a conservation score of 7 out of 9 and describing it as a highly conserved, exposed and buried residue based on the results of the NACSES algorithm (Figure 3). Additionally, the phastCons (reflect the probability of the conservation element, based on the multiple alignments of genome sequences of 46 different species; values range: 0-1) and phyloP (separately measures conservation, ignoring the effects of their neighbors; values range: Between -14 and +6) were used to determine the level of conservation (Table 2). The average conservation grades were 1.00 and 6.09 based on phastCons and phyloP analysis, respectively. The multiple simulations predicted that the amino acid sequence would change and protein features would be altered due to a cysteine’s replacement of the highly conserved amino acid glycine in the catalytic domain, resulting in decreased catalytic activity and loss of calpain-3 catalytic function. Therefore, missense substitution at this position appears to have significant muscular effects, possibly resulting in calpainopathies and muscular dystrophies.
The pathogenicity of the identified variant was also predicted and evaluated by different tools, all of which were unanimous on the “pathogenic variant”. The NM_000070.3:c.661G>T variant was classified as “pathogenic, class 1” based on the following 2015 ACMG rules: PVS1, PM2, PP3, and PP4. According to the in silico and prediction tools, PolyPhen, SIFT, and MutationTaster, this missense variant was predicted to be damaging (PolyPhen2_HDIV: 1.00, D), deleterious (SIFT: 0.00, D), and disease-causing (MutationTaster69: 1.00, D) (Table 2). We also used Pathogenic Mutation Prediction (PMut) analysis, which predicts all missense variations in the protein to predict our variant’s pathogenicity score. Based on PMut analysis, all substitutions at p.221 of the CAPN3 protein were pathogenic, including the glycine to cysteine substitution, which was predicted as a “disease” with a prediction score of 0.79 out of 1.00 (79%).
4. Discussion
LGMDs are a group of genetically heterogeneous diseases characterized by progressive muscle weakness, predominantly affecting the voluntary muscles around the hips and shoulders. The prevalence of LGMDs is estimated to be around 1 in 14500 to 1 in 123000 individuals worldwide (Chen et al., 2021; Pozsgai et al., 2021). LGMDs are caused by mutations in at least 35 genes, each of which can result in a distinct subtype of the LGMDs. One of the subtypes of LGMD, known as LGMD2A or calpainopathy, is caused by mutations in the CAPN3 gene. The gene is responsible for encoding the calpain-3 protein, which plays a crucial role in maintaining the structure and function of muscle fibers. Mutations in the CAPN3 gene could lead to reduced or absent calpain-3 protein, which results in progressive muscle weakness. CAPN3 mutations are one of the most common causes of LGMD, accounting for 20%-30% of all cases of LGMD worldwide, and around 500 unique pathogenic variants, the majority of which are CAPN3 missense mutations (50%–60%), have been found so far associated with LGMD (Park et al., 2016; Zhang et al., 2021). The CAPN3 transcript variant-1 gene (ENST00000397163.8, NM_000070.3) is responsible for encoding the isoform-I of calpain-3. This particular member of the calpain large subunit family exhibits a high affinity for binding to titin, specifically within muscle tissues (Ermolova et al., 2011). It is required for calcium uptake by the sarcoplasmic reticulum and is essential for controlling sarcomeres’ formation, maintenance, and remodeling (Michel et al., 2016). The calpain protein exhibits a structural organization of four distinct domains, with a Penta-EF hand motif in the C-terminal region (Figure 1). Upon binding with calcium ions (Ca2+), domains I and II exhibit cysteine proteases’ enzymatic activity characteristics, forming the Cys Pc domains, PC1 and PC2 (Keira et al., 2003; Michel et al., 2016). Domain III encompasses a pair of calmodulin-binding sites, commonly called a CBSW (calpain-type b-sandwich) domain. Calpain-3 can be distinguished from other calpains involved in autocatalytic activity by the presence of two specific insertional sequences, IS1 and IS2 (Ermolova et al., 2015). Utilizing domain IV’s PEF sequences, calpain-3 undergoes homodimerization, resulting in the orientation of the two catalytic domains in opposite directions. The activity of calpain-3 necessitates binding Ca2+ and Na+ ions (Fanin et al., 2007).
The CAPN3 mutations identified in LGMD2A patients include missense, nonsense, frameshift, splice site, and large deletions or duplications. Most CAPN3 mutations are unique to individual families, although some variations have been reported in multiple families or populations. Moreover, the frequency, progression, clinical, and pathological features of cases described within the same family are variable, even among individuals with the same mutations. Some variations inhibit proteolytic activity, while others interfere with the interaction with substrates or binding targets. Therefore, each variant might lead to diverse consequences in the calpain-3 protein sequence, consequently causing disruptions in its function. Common symptoms include progressive muscle weakness, wasting in the pelvic and shoulder girdles, difficulty walking, and respiratory insufficiency. Accurate LGMD diagnosis is challenging because of the distinct subtypes’ genetic variability and clinical similarities (Zheng et al., 2021). Identifying three autosomal recessive subtypes associated with CAPN3 mutations can be accomplished by examining muscle weakness distribution and age of onset. These subtypes include leiden mobius LGMD (shoulder- and limb-girdle psoas muscular dystrophy), Erb LGMD (shoulder and bone LGMD), and hyper-CK-emia, frequently observed in children and young adults (Dorobek et al., 2015). In most cases, individuals affected by this condition exhibit no muscular symptoms, but they may be diagnosed with elevated levels of serum CK (Fanin & Angelini, 2015). Muscle dystrophy can also be diagnosed using EMG, which reveals the electrical activity of the muscle. Nevertheless, neither method can determine precisely the type of muscle disorder (Pegoraro & Hoffman, 1993; Ten Dam et al., 2019). Fortunately, the continuous progress in genomic technology has facilitated the emergence of novel diagnostic approaches, exemplified by the advent of WES. With the development of molecular testing technologies, we can now employ WES as a fast and accurate tool for diagnosing and detecting the causative mutations of muscular dystrophies. This technique exhibits numerous advantages compared to alternative methodologies employed for diagnosing LGMD. Savarese et al. (2016) observed that WES demonstrated a genetic etiology for LGMD in 47% of the examined patients. This percentage was notably higher than the diagnostic yield of 31% achieved through conventional diagnostic approaches. The utilization of WES has demonstrated the capacity to identify previously uncharacterized mutations that have not been previously linked to LGMD or other muscular disorders. This phenomenon can facilitate the identification of novel genes associated with disease and serve as a basis for identifying potential targets for therapeutic interventions. In addition, the WES procedure exhibits notable efficiency regarding turnaround time, as it can be executed expeditiously, yielding outcomes that become accessible within a few weeks (Narasimhaiah et al., 2022). Therefore, it could facilitate expedited identification and intervention for individuals afflicted with LGMD.
Nevertheless, it is imperative to acknowledge the existence of certain constraints associated with WES. These factors encompass the financial implications of the test, the possibility of unexpected discoveries, and the requirement for specialized knowledge to analyze and comprehend the outcomes accurately. Notwithstanding these constraints, WES has emerged as a valuable instrument in diagnosing LGMD and other genetic disorders.
In this research, we looked at a family with two affected offspring strongly suspected of LGMD born to a consanguineous spouse. We applied WES, followed by segregation analysis in family members, to pinpoint the causative mutations. The proband’s exome revealed a pathogenic CAPN3 variant, NM_000070.3:c.661G>T, not previously reported in LGMD patients as a solitary variant. This variant was also observed in a second afflicted family member and the parents in the homozygous and heterozygous states. The c.661G>T:p.Gly221Cys variant causes a substitution of the amino acid glycine at position 221 with cysteine in the calpain-3 protein sequence. This change occurs in the protease domain of the protein, which is responsible for its enzymatic activity. The substitution of glycine with cysteine introduces a new sulfur-containing group into the protein structure, which can form disulfide bonds with other cysteine residues. Glycine is classified as a compact and pliable amino acid, capable of accommodating itself within constricted regions of protein structures. Conversely, cysteine exhibits a comparatively larger and less flexible nature, enabling it to establish disulfide linkages with other cysteine residues. Consequently, p.Gly221Cys may disrupt the protein’s structure and interfere with its ability to carry out normal physiological functions.
The identified CAPN3 variant is absent from public databases, and there are no reports of this mutation concerning LGMD in the HGMD, dbSNP, gnomAD, 1000 Genomes Project Database, Ensembl, ESP, and ExAC. The present investigation is the first to describe this pathogenic mutation in both homozygous and heterozygous forms in a family with LGMD, highlighting its pathogenicity potential. Notably, just one report of this variation in a case has been clinically recorded in ClinVar, and the resulting condition has been described as an “abnormality of the muscles.” Consequently, we submitted and indexed the missense variant observed in this investigation as the first report in patients with LGMD in the ClinVar database under the accession number RCV002512157.1.
5. Conclusion
In the current study, we demonstrated the relevance of WES in identifying a pathogenic variant as a cause of muscular dystrophy and reported for the first time a homozygous CAPN3 mutation segregated in a family with LGMD2A. Genetic counseling and personalized medicine rely on discovering novel genes or their variations. We infer that the c.661G>T:p.Gly221Cys variation in exon 5 of the CAPN3 gene alters the normal function of the calpain-3 protein domain, hence impairing the protein’s enzymatic activity. However, further research, including a functional examination and cell models, is required to substantiate this variation.
The discovery of a novel pathogenic variant in the CAPN3 gene, which causes LGMD2A, expands the genetic spectrum of this rare and heterogeneous group of muscular disorders. The functional consequences of this variant on the structure and activity of the calpain-3 protein remain to be elucidated. Further studies are needed to investigate how the p.Gly221Cys substitution affects the protease domain of the protein and how this impacts the cellular processes that involve calpain-3, such as muscle regeneration, calcium signaling, and cytoskeletal remodeling. Moreover, the absence of this variant in public databases and its pathogenicity potential, as evidenced by its presence in a family with LGMD, underscores the need for future studies to explore its specific impact on protein/cellular function. Understanding the functional consequences of this variant is crucial for developing targeted therapeutic interventions and personalized therapeutic strategies for LGMD patients.
Ethical Considerations
Compliance with ethical guidelines
The research project was approved by the North Tehran Branch, Islamic Azad University, Tehran, Iran (Code IR.IAU.TNB.REC.1400.080; dated 2021-11-07). All procedures performed in the studies involving human participants were in accordance with the ethical standards of the institutional and or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. All experiments were carried out under the ethical standards of the relevant institutional committees. After that, all participants in this research received informed consent to publish any potentially identifying data in this paper.
Funding
This paper was extracted from the PhD dissertation of Amin Sakhaei, approved by the Department of Biology, Faculty of Biological Sciences, North Tehran Branch, Islamic Azad University, Tehran, Iran. This research was financially supported by a grant from the Vice-Chancellor for Research Affairs of North Tehran Branch, Islamic Azad University, Tehran, Iran.
Authors' contributions
Conceptualization, methodology, investigation, writing the original draft: Amin Sakhaei; Supervision, funding administration data analysis, review and editing: Javad Mohammadi-Asl; Data collection: All authors. All authors have read and approved the manuscript’s content and confirmed the accuracy or integrity of any part of the work.
Conflict of interest
The authors declared no conflict of interest.
Acknowledgments
The authors thank their colleagues in the Department of Biology, Faculty of Biological Sciences, North Tehran Branch, Islamic Azad University, Tehran, Iran. The authors appreciate the coworkers and patients who kindly contributed to this study. The authors are especially thankful to the personnel of the Noorgene Genetic and Clinical Laboratory, Ahvaz, Iran.
References
Clinically and genetically, limb-girdle muscular dystrophies (LGMDs) are a diverse group of muscle abnormalities that can be passed down in an autosomal recessive or dominant pattern (Bockhorst & Wicklund, 2020). On clinical examination, patients have symmetrical weakness in the pelvic, scapular, and trunk muscles. According to evidence, over 30 different genetic subtypes of LGMD have been discovered (Straub et al., 2018). Autosomal recessive limb-girdle muscular dystrophy-1 (LGMDR1; MIM:253600; ORPHA:267), also referred to as limb-girdle muscular dystrophy type 2A (LGMD2A), is the most common form of LGMD characterized by the progressive weakness of the proximal limb muscles and elevated serum creatine kinase (CK) levels (Nascimbeni et al., 2010). The evidence indicates that LGMDR1 is the first human disease discovered to be caused by mutations in the calcium-activated neutral proteinase 3 (CAPN3) gene (Chro. 15q15.1, OMIM: 114240). The gene encodes the calpain-3 protein, accounting for about 30% of all LGMD cases (Martinez-Thompson et al., 2018; Sorimachi et al., 2010). Although the facial and neck muscles are unaffected, the clinical features of LGMD2A are highly heterogeneous, present at any age (from infancy to adulthood), and include running difficulties, a waddling gait, scapular winging, as well as respiratory failure in the advanced stages of the disease (Hunter et al., 2019; Pozsgai et al., 2021). LGMD cases involving cardiac muscles have also been reported sporadically (Mori-Yoshimura et al., 2017; Narayanaswami et al., 2014). The CAPN3 gene product (non-lysosomal Ca2+-dependent cysteine protease), which consists of four domains (protease core subdomain-1, protease core subdomain-2, C2 domain-like domain, and penta-EF-hand domain) and 821 amino acids, cleaves and breaks down numerous essential skeletal myoproteins, particularly those banded with the structure of myofibrils (Ono et al., 2016) (Figure 1). This breakdown is vital for muscle maintenance and regeneration, as it allows for removing damaged or old proteins and replacing them with new ones. Calpain-3 also forms and matures muscle fibers during development and regulates muscle contraction. Physiological aberrations caused by CAPN3 defects can result in insufficient ryanodine regulation, leading to movement anomalies and muscular weakness (Taghizadeh et al., 2019).
Based on the Human Gene Mutation Database (HGMD) and Leiden Open Variation Database (LOVD), more than 500 different CAPN3 variants have been identified in LGMD cases. Investigations indicate that missense variants are the most typical, comprising more than half of all CAPN3 mutations and reported as mainly compound heterozygous (Fanin et al., 2014; Park et al., 2016). Each variant affects different amino acids and can cause diverse consequences in the calpain-3 protein sequence, disrupting its function. Many patients may be misdiagnosed in the early stages due to the high clinical and genetic heterogeneity of LGMDs. Effective genetic diagnosis is required for proper genetic counseling and treatment of LGMD patients (Taghizadeh et al., 2019). Because of the wide range of LGMD-causing variants, next-generation sequencing (NGS) is one of the best options for the conclusive differential diagnosis of LGMD (Fanin et al., 2009; Piluso et al., 2005). However, the causal mutation for more than half of LGMD cases remains unknown despite recent advancements in molecular diagnostic techniques (Angelini, 2020). In the current study, whole-exome sequencing (WES) was utilized to detect LGMD-causing variants in the proband, followed by Sanger sequencing to confirm and segregate the detected variation in the proband and the family members. The WES results revealed a pathogenic homozygous variant within the CAPN3 gene in the proband, which was confirmed and co-segregated with LGMD2A in the family. This study is the first report of this variant in LGMD cases, enriching our knowledge concerning LGMD pathology and facilitating the future clinical management of these patients.
2. Materials and Methods
Electroneurographic, laboratory, and clinical findings
A consanguineous couple with two affected offspring strongly suspected of LGMD was referred to our Genetic Department at Ahvaz Noorgene Genetic and Clinical Laboratory to diagnose their muscular dystrophy type. The parents were first cousins, with two affected and three healthy offspring (Figure 2). The proband, a 33-year-old woman, was the oldest affected member in this family with a history of progressive muscular weakness that started at age 13. Her younger brother, a 27-year-old man, was another affected member whose progressive muscle weakness appeared since 16. Both affected had transient growing pains, sit-to-stand difficulties, a waddling gait, and mild proximal muscle weakness without involving facial muscles. Neither had intellectual disabilities, but calf pseudohypertrophy was present only in the proband. Though neither parent showed any symptoms of muscular weakness, the father was undergoing treatment for bladder cancer during the study. Additionally, the proband was getting therapy for an ovarian cyst. LGMD was suggested to be the first-line diagnosis by clinicians based on clinical presentations, electromyography (EMG), and laboratory tests.
The proband’s evoked EMG and nerve conduction velocity (NCV) examinations at the age of 13 demonstrated short-duration motor units, early recruitment, and wasting of the deltoids bilaterally with 2+ fibrillations. Electroneurography revealed the significant weakness of deltoids (grade 2/5) and positive sharp waves at rest with a brief duration (3-6 ms, NCV: 33 m/s). Laboratory parameters, including serum potassium, erythrocyte sedimentation rate, and C-reactive protein level, were all within normal ranges. However, the creatinine phosphokinase level was significantly elevated at 1187 U/L (reference range: 24-170 U/L). We applied WES to investigate the proband’s exome and identify the causal mutations in this LGMD2A-suspected family.
WES
After providing the proband and his family with an explanation of the NGS procedure, informed consent was obtained from them. Genomic DNA was isolated from the whole blood of the proband and all family members using a salting-out standard protocol in the presence of ethylenediaminetetraacetic acid (EDTA) as an anticoagulant. A NanoDrop200 spectrophotometer was used to determine the gDNA’s optical density (Thermo Fisher Scientific, Inc.). Exon capture and enrichment were conducted on genomic DNA using the Agilent SureSelectXT Human All Exon V8 (Agilent, Santa Clara, CA), and whole coding areas were sequenced with an average depth of 100x on an Illumina HiSeq 2000 sequencer (Illumina, Inc., San Diego, CA, USA) at the Noorgene Genetic and Clinical Laboratory. The raw data of WES was evaluated using a Core i7 central processing unit and the Linux (Ubuntu 20.04) operating system. As detailed below, various in silico prediction methods and bioinformatics resources were employed to provide additional support for the identified variants:
Quality control of the read lengths was evaluated by the FastQC program (version 0.11.9) based on a GC content of 50% and a Phred score of 20. Trimmomatic software (version 0.36) was used to eliminate adapter-contaminated and low-quality readings.
The Burrows–Wheeler Aligner (BWA) aligning tool (version 2.4.0) was used to precisely map sequence reads to the human reference genome (GRCh38/hg19), and outputs were generated in sequence alignment/map (SAM) file format. Afterward, the SAM file was converted to a binary alignment map file using the Picard program.
The local realignment of insertion/deletions (indels), base quality score recalibration, single-nucleotide variations (SNVs)/indels calling, and data compression were accomplished using a combination of genome analysis tool HaplotypeCaller and VarScan mpileup2snp protocol.
Gene ontology reference genome project, multiple association network integration method, protein analysis through evolutionary relationships, and Ensembl variant effect predictor (VEP) were used to annotate the output VCF file (Ensembl VEP).
Using the R programming command-line program, the annotated variations were filtered in the following ways.
Databases dbSNP v137, 1000 genomes project, Exome Sequencing Project, Human Gene Mutation Database (HGMD), Genome Aggregation Database (gnomAD), Iranome, and the Exome Aggregation Consortium (ExAC) were used to annotate the minor allele frequencies of all known variations.
Utilizing CADD (combined annotation-dependent depletion), PHRE, and GERP (the genomic evolutionary rate profiling) scores, we could filter out synonymous, deeply intronic, or less conserved variations.
Using the Phenolyzer (phenotype-based prioritization) tool and the Human Phenotype Ontology (HPO), we could separate the pathogenic variants from the non-pathogenic ones based on the presented clinical/paraclinical results.
According to standards established by the American College of Medical Genetics and Genomics Association for Molecular Pathology (ACMG/AMP), the remaining variations were analyzed and categorized. Besides, pathogenicity prediction of varieties was carried out through different bioinformatic tools: PolyPhen-2, SIFT, Pathogenic Mutation Prediction (PMut), and the MutationTaster.
Finally, the Integrative Genomics Viewer (IGV) program scrutinized the depth of reads for each variation.
Confirmation of variant and family segregation
The variation discovered through WES was confirmed by Sanger sequencing using an ABI3130xl sequencer and a BigDye Terminator v.3.1 Cycle Sequencing Kit (Life Technologies, USA), which also allowed for the investigation of family segregation. Primer3 web-based software was used to design the primer sequences spanning the WES-identified variation in the proband. Both the forward-primer (CAPN3-EX5-F278: 5′-ATT GTT TCC ATC CCA TGA GC-3′) and the reverse-primer (CAPN3-EX5-R278: 5′-TGA GAA ATT CCC AGT CCT CAA-3′) were put through the OligoAnalyzer tool to investigate their properties and evaluate their quality. Following the PCR amplification for the area containing the WES-identified variation, Sanger sequencing was conducted to confirm the potential causal variant in the proband and her family members.
The targeted area was amplified by running a PCR reaction in a total volume of 25 μL comprising 250 nM dNTPs, 100 ng of template DNA, 0.5 mM of each primer, and 1.25 units AmpliTaq Gold DNA polymerase in 1×reaction buffer on a FlexCycler Thermocycler (10 mM Tris HCl, pH 8.3, 50 mM KCl, 2.5 mM MgCl2). PCR procedure was conducted as follows: Initial denaturation at 95 ⁰C for 3 minutes, followed by 35 cycles of denaturation at 95 ⁰C for 45 seconds, annealing at 58 ⁰C for 1 minute, and extension at 72 ⁰C for 45 seconds, followed by a final 5-minute extension at 72 ⁰C. Purified PCR products were sequenced using a BigDye Terminator v.3.1 Cycle Sequencing Kit with inclined primers and PCR conditions. CodonCode aligner software was utilized to assemble and align Sanger sequencing files (version 9.0.1).
3. Results
WES data and co-segregation analysis
Variants within genes known to be involved in muscular dystrophies were prioritized for WES data analysis in this study. WES data were filtered to exclude non-genetic variants and compared with the databases. Following the WES data analysis of known muscular dystrophy genes with a recessive inheritance pattern and in silico analysis, we discovered a pathogenic homozygous variant within the CAPN3 gene, NM_000070.3:exon 5:c.661>GT, that had not previously been reported to the LGMD phenotypes in public databases (Table 1).
As shown in Figure 3, glycine at position 221 is highly conserved across species, emphasizing its critical role in protein function. The substitution of glycine with cysteine in this position disrupts the protein’s structure and function.
Subsequently, the pathogenic CAPN3 variant was validated through Sanger sequencing and segregated with LGMDA2 in the proband’s family (Figure 4). The WES and Sanger sequencing results consistently revealed that a GGT>TGT substitution at c.661 in exon 5 of the proband’s coding sequence (CDS) exists as a single nucleotide variant (SNV), and he was homozygous for this variant (TGT/TGT). Co-segregation analysis indicated that the proband’s brother was also homozygous for this variation (TGT/TGT), and both afflicted offspring had heterozygous carrier parents (GGT/TGT). In contrast, the healthy siblings did not carry the variant and were homozygous for the wild type (G) in the NM_000070.3:c.661 (GGT/GGT). This variation has not been reported as associated with LGMD in databases such as the HGMD, ESP, ExAC browser, LOVD, and dbSNP. In conclusion, the CAPN3 variation was segregated with LGMDA2 in this family, suggesting it could be considered the proband disease-causing.
Variant analysis
The missense variant c.661G>T:p.Gly221Cys resulted in a single nucleotide alternation (G>T) at site 661 in the coding region of CAPN3 (NM_000070.3), which introduced a glycine (Gly: [G]) residue that replaced a cysteine (Cys: [C]) at position 221 of the protein sequence (Table 1). Multiple sequence alignment analysis of the CAPN3 protein revealed that p.Gly221 is a highly conserved amino acid across many species (tfbsConsSites-score:940, phyloP100way_vertebrate: 9.905, phyloP20way_mammalian: 1.044, SiPhy_29way_logOdds: 19.398) (Table 2). The detected variant is topologically located in the peptidase_C2 domain, a highly conserved region, emphasizing the importance of the changed amino acid (glycine to cysteine) in protein function (Figure 3). The I-Mutant2.0 server, which computes the ΔΔG values of variations (DG “new protein”- DG “wildtype”), determined that the p.Gly221Cys substitution reduces the stability of the CAPN3 protein (ΔΔG=-0.55 kcal/mol, ΔΔG <0: Decrease stability). The ConSurf service also ran a conservation analysis on G.221, reporting a conservation score of 7 out of 9 and describing it as a highly conserved, exposed and buried residue based on the results of the NACSES algorithm (Figure 3). Additionally, the phastCons (reflect the probability of the conservation element, based on the multiple alignments of genome sequences of 46 different species; values range: 0-1) and phyloP (separately measures conservation, ignoring the effects of their neighbors; values range: Between -14 and +6) were used to determine the level of conservation (Table 2). The average conservation grades were 1.00 and 6.09 based on phastCons and phyloP analysis, respectively. The multiple simulations predicted that the amino acid sequence would change and protein features would be altered due to a cysteine’s replacement of the highly conserved amino acid glycine in the catalytic domain, resulting in decreased catalytic activity and loss of calpain-3 catalytic function. Therefore, missense substitution at this position appears to have significant muscular effects, possibly resulting in calpainopathies and muscular dystrophies.
The pathogenicity of the identified variant was also predicted and evaluated by different tools, all of which were unanimous on the “pathogenic variant”. The NM_000070.3:c.661G>T variant was classified as “pathogenic, class 1” based on the following 2015 ACMG rules: PVS1, PM2, PP3, and PP4. According to the in silico and prediction tools, PolyPhen, SIFT, and MutationTaster, this missense variant was predicted to be damaging (PolyPhen2_HDIV: 1.00, D), deleterious (SIFT: 0.00, D), and disease-causing (MutationTaster69: 1.00, D) (Table 2). We also used Pathogenic Mutation Prediction (PMut) analysis, which predicts all missense variations in the protein to predict our variant’s pathogenicity score. Based on PMut analysis, all substitutions at p.221 of the CAPN3 protein were pathogenic, including the glycine to cysteine substitution, which was predicted as a “disease” with a prediction score of 0.79 out of 1.00 (79%).
4. Discussion
LGMDs are a group of genetically heterogeneous diseases characterized by progressive muscle weakness, predominantly affecting the voluntary muscles around the hips and shoulders. The prevalence of LGMDs is estimated to be around 1 in 14500 to 1 in 123000 individuals worldwide (Chen et al., 2021; Pozsgai et al., 2021). LGMDs are caused by mutations in at least 35 genes, each of which can result in a distinct subtype of the LGMDs. One of the subtypes of LGMD, known as LGMD2A or calpainopathy, is caused by mutations in the CAPN3 gene. The gene is responsible for encoding the calpain-3 protein, which plays a crucial role in maintaining the structure and function of muscle fibers. Mutations in the CAPN3 gene could lead to reduced or absent calpain-3 protein, which results in progressive muscle weakness. CAPN3 mutations are one of the most common causes of LGMD, accounting for 20%-30% of all cases of LGMD worldwide, and around 500 unique pathogenic variants, the majority of which are CAPN3 missense mutations (50%–60%), have been found so far associated with LGMD (Park et al., 2016; Zhang et al., 2021). The CAPN3 transcript variant-1 gene (ENST00000397163.8, NM_000070.3) is responsible for encoding the isoform-I of calpain-3. This particular member of the calpain large subunit family exhibits a high affinity for binding to titin, specifically within muscle tissues (Ermolova et al., 2011). It is required for calcium uptake by the sarcoplasmic reticulum and is essential for controlling sarcomeres’ formation, maintenance, and remodeling (Michel et al., 2016). The calpain protein exhibits a structural organization of four distinct domains, with a Penta-EF hand motif in the C-terminal region (Figure 1). Upon binding with calcium ions (Ca2+), domains I and II exhibit cysteine proteases’ enzymatic activity characteristics, forming the Cys Pc domains, PC1 and PC2 (Keira et al., 2003; Michel et al., 2016). Domain III encompasses a pair of calmodulin-binding sites, commonly called a CBSW (calpain-type b-sandwich) domain. Calpain-3 can be distinguished from other calpains involved in autocatalytic activity by the presence of two specific insertional sequences, IS1 and IS2 (Ermolova et al., 2015). Utilizing domain IV’s PEF sequences, calpain-3 undergoes homodimerization, resulting in the orientation of the two catalytic domains in opposite directions. The activity of calpain-3 necessitates binding Ca2+ and Na+ ions (Fanin et al., 2007).
The CAPN3 mutations identified in LGMD2A patients include missense, nonsense, frameshift, splice site, and large deletions or duplications. Most CAPN3 mutations are unique to individual families, although some variations have been reported in multiple families or populations. Moreover, the frequency, progression, clinical, and pathological features of cases described within the same family are variable, even among individuals with the same mutations. Some variations inhibit proteolytic activity, while others interfere with the interaction with substrates or binding targets. Therefore, each variant might lead to diverse consequences in the calpain-3 protein sequence, consequently causing disruptions in its function. Common symptoms include progressive muscle weakness, wasting in the pelvic and shoulder girdles, difficulty walking, and respiratory insufficiency. Accurate LGMD diagnosis is challenging because of the distinct subtypes’ genetic variability and clinical similarities (Zheng et al., 2021). Identifying three autosomal recessive subtypes associated with CAPN3 mutations can be accomplished by examining muscle weakness distribution and age of onset. These subtypes include leiden mobius LGMD (shoulder- and limb-girdle psoas muscular dystrophy), Erb LGMD (shoulder and bone LGMD), and hyper-CK-emia, frequently observed in children and young adults (Dorobek et al., 2015). In most cases, individuals affected by this condition exhibit no muscular symptoms, but they may be diagnosed with elevated levels of serum CK (Fanin & Angelini, 2015). Muscle dystrophy can also be diagnosed using EMG, which reveals the electrical activity of the muscle. Nevertheless, neither method can determine precisely the type of muscle disorder (Pegoraro & Hoffman, 1993; Ten Dam et al., 2019). Fortunately, the continuous progress in genomic technology has facilitated the emergence of novel diagnostic approaches, exemplified by the advent of WES. With the development of molecular testing technologies, we can now employ WES as a fast and accurate tool for diagnosing and detecting the causative mutations of muscular dystrophies. This technique exhibits numerous advantages compared to alternative methodologies employed for diagnosing LGMD. Savarese et al. (2016) observed that WES demonstrated a genetic etiology for LGMD in 47% of the examined patients. This percentage was notably higher than the diagnostic yield of 31% achieved through conventional diagnostic approaches. The utilization of WES has demonstrated the capacity to identify previously uncharacterized mutations that have not been previously linked to LGMD or other muscular disorders. This phenomenon can facilitate the identification of novel genes associated with disease and serve as a basis for identifying potential targets for therapeutic interventions. In addition, the WES procedure exhibits notable efficiency regarding turnaround time, as it can be executed expeditiously, yielding outcomes that become accessible within a few weeks (Narasimhaiah et al., 2022). Therefore, it could facilitate expedited identification and intervention for individuals afflicted with LGMD.
Nevertheless, it is imperative to acknowledge the existence of certain constraints associated with WES. These factors encompass the financial implications of the test, the possibility of unexpected discoveries, and the requirement for specialized knowledge to analyze and comprehend the outcomes accurately. Notwithstanding these constraints, WES has emerged as a valuable instrument in diagnosing LGMD and other genetic disorders.
In this research, we looked at a family with two affected offspring strongly suspected of LGMD born to a consanguineous spouse. We applied WES, followed by segregation analysis in family members, to pinpoint the causative mutations. The proband’s exome revealed a pathogenic CAPN3 variant, NM_000070.3:c.661G>T, not previously reported in LGMD patients as a solitary variant. This variant was also observed in a second afflicted family member and the parents in the homozygous and heterozygous states. The c.661G>T:p.Gly221Cys variant causes a substitution of the amino acid glycine at position 221 with cysteine in the calpain-3 protein sequence. This change occurs in the protease domain of the protein, which is responsible for its enzymatic activity. The substitution of glycine with cysteine introduces a new sulfur-containing group into the protein structure, which can form disulfide bonds with other cysteine residues. Glycine is classified as a compact and pliable amino acid, capable of accommodating itself within constricted regions of protein structures. Conversely, cysteine exhibits a comparatively larger and less flexible nature, enabling it to establish disulfide linkages with other cysteine residues. Consequently, p.Gly221Cys may disrupt the protein’s structure and interfere with its ability to carry out normal physiological functions.
The identified CAPN3 variant is absent from public databases, and there are no reports of this mutation concerning LGMD in the HGMD, dbSNP, gnomAD, 1000 Genomes Project Database, Ensembl, ESP, and ExAC. The present investigation is the first to describe this pathogenic mutation in both homozygous and heterozygous forms in a family with LGMD, highlighting its pathogenicity potential. Notably, just one report of this variation in a case has been clinically recorded in ClinVar, and the resulting condition has been described as an “abnormality of the muscles.” Consequently, we submitted and indexed the missense variant observed in this investigation as the first report in patients with LGMD in the ClinVar database under the accession number RCV002512157.1.
5. Conclusion
In the current study, we demonstrated the relevance of WES in identifying a pathogenic variant as a cause of muscular dystrophy and reported for the first time a homozygous CAPN3 mutation segregated in a family with LGMD2A. Genetic counseling and personalized medicine rely on discovering novel genes or their variations. We infer that the c.661G>T:p.Gly221Cys variation in exon 5 of the CAPN3 gene alters the normal function of the calpain-3 protein domain, hence impairing the protein’s enzymatic activity. However, further research, including a functional examination and cell models, is required to substantiate this variation.
The discovery of a novel pathogenic variant in the CAPN3 gene, which causes LGMD2A, expands the genetic spectrum of this rare and heterogeneous group of muscular disorders. The functional consequences of this variant on the structure and activity of the calpain-3 protein remain to be elucidated. Further studies are needed to investigate how the p.Gly221Cys substitution affects the protease domain of the protein and how this impacts the cellular processes that involve calpain-3, such as muscle regeneration, calcium signaling, and cytoskeletal remodeling. Moreover, the absence of this variant in public databases and its pathogenicity potential, as evidenced by its presence in a family with LGMD, underscores the need for future studies to explore its specific impact on protein/cellular function. Understanding the functional consequences of this variant is crucial for developing targeted therapeutic interventions and personalized therapeutic strategies for LGMD patients.
Ethical Considerations
Compliance with ethical guidelines
The research project was approved by the North Tehran Branch, Islamic Azad University, Tehran, Iran (Code IR.IAU.TNB.REC.1400.080; dated 2021-11-07). All procedures performed in the studies involving human participants were in accordance with the ethical standards of the institutional and or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. All experiments were carried out under the ethical standards of the relevant institutional committees. After that, all participants in this research received informed consent to publish any potentially identifying data in this paper.
Funding
This paper was extracted from the PhD dissertation of Amin Sakhaei, approved by the Department of Biology, Faculty of Biological Sciences, North Tehran Branch, Islamic Azad University, Tehran, Iran. This research was financially supported by a grant from the Vice-Chancellor for Research Affairs of North Tehran Branch, Islamic Azad University, Tehran, Iran.
Authors' contributions
Conceptualization, methodology, investigation, writing the original draft: Amin Sakhaei; Supervision, funding administration data analysis, review and editing: Javad Mohammadi-Asl; Data collection: All authors. All authors have read and approved the manuscript’s content and confirmed the accuracy or integrity of any part of the work.
Conflict of interest
The authors declared no conflict of interest.
Acknowledgments
The authors thank their colleagues in the Department of Biology, Faculty of Biological Sciences, North Tehran Branch, Islamic Azad University, Tehran, Iran. The authors appreciate the coworkers and patients who kindly contributed to this study. The authors are especially thankful to the personnel of the Noorgene Genetic and Clinical Laboratory, Ahvaz, Iran.
References
Angelini, C. (2020). LGMD. Identification, description and classification. Acta Myologica, 39(4), 207-217. [DOI:10.36185/2532-1900-024]
Bockhorst, J., & Wicklund, M. (2020). Limb Girdle Muscular Dystrophies. Neurologic Clinics, 38(3), 493–504. [DOI:10.1016/j.ncl.2020.03.009] [PMID]
Chen, L., Tang, F., Gao, H., Zhang, X., Li, X., & Xiao, D. (2021). CAPN3: A musclespecific calpain with an important role in the pathogenesis of diseases (Review). International Journal of Molecular Medicine, 48(5), 203. [DOI:10.3892/ijmm.2021.5036] [PMID]
Dorobek, M., Ryniewicz, B., Kabzińska, D., Fidziańska, A., Styczyńska, M., & Hausmanowa-Petrusewicz, I. (2015). The Frequency of c.550delA Mutation of the CANP3 Gene in the Polish LGMD2A population. Genetic Testing and Molecular Biomarkers, 19(11), 637–640. [DOI:10.1089/gtmb.2015.0131] [PMID]
Ermolova, N., Kramerova, I., & Spencer, M. J. (2015). Autolytic activation of calpain 3 proteinase is facilitated by calmodulin protein. The Journal of Biological Chemistry, 290(2), 996–1004. [DOI:10.1074/jbc.M114.588780] [PMID]
Ermolova, N., Kudryashova, E., DiFranco, M., Vergara, J., Kramerova, I., & Spencer, M. J. (2011). Pathogenity of some limb girdle muscular dystrophy mutations can result from reduced anchorage to myofibrils and altered stability of calpain 3. Human Molecular Genetics, 20(17), 3331–3345. [DOI:10.1093/hmg/ddr239] [PMID]
Fanin, M., & Angelini, C. (2015). Protein and genetic diagnosis of limb girdle muscular dystrophy type 2A: The yield and the pitfalls. Muscle & Nerve, 52(2), 163–173. [DOI:10.1002/mus.24682] [PMID]
Fanin, M., Nascimbeni, A. C., & Angelini, C. (2007). Screening of calpain-3 autolytic activity in LGMD muscle: A functional map of CAPN3 gene mutations. Journal of Medical Genetics, 44(1), 38–43.[DOI:10.1136/jmg.2006.044859] [PMID]
Fanin, M., Nascimbeni, A. C., & Angelini, C. (2014). Gender difference in limb-girdle muscular dystrophy: A muscle fiber morphometric study in 101 patients. Clinical Neuropathology, 33(3), 179–185. [DOI:10.5414/np300728] [PMID]
Fanin, M., Nascimbeni, A. C., Tasca, E., & Angelini, C. (2009). How to tackle the diagnosis of limb-girdle muscular dystrophy 2A. European Journal of Human Genetics: EJHG, 17(5), 598–603. [DOI:10.1038/ejhg.2008.193] [PMID]
Hunter, M., Heatwole, C., Wicklund, M., Weihl, C. C., Mozaffar, T., & Statland, J. M., et al. (2019). Limb-girdle muscular dystrophy: A perspective from adult patients on what matters most. Muscle & Nerve, 60(4), 419–424. [DOI:10.1002/mus.26636] [PMID]
Keira, Y., Noguchi, S., Minami, N., Hayashi, Y. K., & Nishino, I. (2003). Localization of calpain 3 in human skeletal muscle and its alteration in limb-girdle muscular dystrophy 2A muscle. Journal of Biochemistry, 133(5), 659–664. [DOI:10.1093/jb/mvg084] [PMID]
Martinez-Thompson, J. M., Moore, S. A., & Liewluck, T. (2018). A novel CAPN3 mutation in late-onset limb-girdle muscular dystrophy with early respiratory insufficiency. Journal of Clinical Neuroscience: Official Journal of The Neurosurgical Society of Australasia, 53, 229–231. [DOI:10.1016/j.jocn.2018.04.025] [PMID]
Michel, L. Y., Hoenderop, J. G., & Bindels, R. J. (2016). Calpain-3-mediated regulation of the Na⁺-Ca²⁺ exchanger isoform 3. Pflugers Archiv: European Journal of Physiology, 468(2), 243–255. [DOI:10.1007/s00424-015-1747-8] [PMID]
Mori-Yoshimura, M., Segawa, K., Minami, N., Oya, Y., Komaki, H., & Nonaka, I., et al. (2017). Cardiopulmonary dysfunction in patients with limb-girdle muscular dystrophy 2A. Muscle & Nerve, 55(4), 465–469. [DOI:10.1002/mus.25369] [PMID]
Narasimhaiah, D., Uppin, M. S., & Ranganath, P. (2022). Genetics and muscle pathology in the diagnosis of muscular dystrophies: An update. Indian Journal of Pathology & Microbiology, 65(Supplement), S259–S270. [PMID]
Narayanaswami, P., Weiss, M., Selcen, D., David, W., Raynor, E., & Carter, G., et al. (2014). Evidence-based guideline summary: Diagnosis and treatment of limb-girdle and distal dystrophies: Report of the guideline development subcommittee of the American Academy of Neurology and the practice issues review panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology, 83(16), 1453-1463. [DOI:10.1212/wnl.0000000000000892] [PMID]
Nascimbeni, A. C., Fanin, M., Tasca, E., & Angelini, C. (2010).Transcriptional and translational effects of intronic CAPN3 gene mutations. Human Mutation, 31(9), E1658–E1669. [DOI:10.1002/humu.21320] [PMID]
Ono, Y., Ojima, K., Shinkai-Ouchi, F., Hata, S., & Sorimachi, H. (2016). An eccentric calpain, CAPN3/p94/calpain-3. Biochimie, 122, 169-187. [DOI:10.1016/j.biochi.2015.09.010] [PMID]
Park, H. J., Jang, H., Lee, J. H., Shin, H. Y., Cho, S. R., & Park, K. D., et al. (2016). Clinical and pathological heterogeneity of Korean patients with CAPN3 mutations. Yonsei Medical Journal, 57(1), 173–179. [DOI:10.3349/ymj.2016.57.1.173] [PMID]
Pegoraro, E., & Hoffman, E. P. (1993). Limb-girdle muscular dystrophy overview - retired chapter, for historical reference only. In M. P. Adam, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. W. Gripp, & A. Amemiya (Eds.). GeneReviews. Seattle: University of Washington. [Link]
Piluso, G., Politano, L., Aurino, S., Fanin, M., Ricci, E., & Ventriglia, V. M., et al. (2005). Extensive scanning of the calpain-3 gene broadens the spectrum of LGMD2A phenotypes. Journal of Medical Genetics, 42(9), 686–693. [DOI:10.1136/jmg.2004.028738] [PMID]
Pozsgai, E., Griffin, D., Potter, R., Sahenk, Z., Lehman, K., & Rodino-Klapac, L. R., et al. (2021). Unmet needs and evolving treatment for limb girdle muscular dystrophies. Neurodegenerative Disease Management, 11(5), 411–429. [DOI:10.2217/nmt-2020-0066] [PMID]
Savarese, M., Di Fruscio, G., Torella, A., Fiorillo, C., Magri, F., & Fanin, M., et al. (2016). The genetic basis of undiagnosed muscular dystrophies and myopathies: Results from 504 patients. Neurology, 87(1), 71-76. [DOI:10.1212/wnl.0000000000002800] [PMID]
Sorimachi, H., Hata, S., & Ono, Y. (2010). Expanding members and roles of the calpain superfamily and their genetically modified animals. Experimental Animals, 59(5), 549–566. [DOI:10.1538/expanim.59.549] [PMID]
Straub, V., Murphy, A., Udd, B., & LGMD workshop study group (2018). 229th ENMC international workshop: Limb girdle muscular dystrophies - Nomenclature and reformed classification Naarden, the Netherlands, 17-19 March 2017. Neuromuscular Disorders: NMD, 28(8), 702–710. [DOI:10.1016/j.nmd.2018.05.007] [PMID]
Taghizadeh, E., Rezaee, M., Barreto, G. E., & Sahebkar, A. (2019). Prevalence, pathological mechanisms, and genetic basis of limb-girdle muscular dystrophies: A review. Journal of Cellular Physiology, 234(6), 7874–7884. [DOI:10.1002/jcp.27907] [PMID]
Ten Dam, L., Frankhuizen, W. S., Linssen, W. H. J. P., Straathof, C. S., Niks, E. H., & Faber, K., et al. (2019). Autosomal recessive limb-girdle and Miyoshi muscular dystrophies in the Netherlands: The clinical and molecular spectrum of 244 patients. Clinical Genetics, 96(2), 126–133. [DOI:10.1111/cge.13544] [PMID]
Type of Study: Original |
Subject:
Cellular and molecular Neuroscience
Received: 2023/12/2 | Accepted: 2024/02/10 | Published: 2025/11/28
Received: 2023/12/2 | Accepted: 2024/02/10 | Published: 2025/11/28
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
