

Volume 16, Issue 1 (January & February 2025)
BCN 2025, 16(1): 107-114 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Shpilyukova Y A, Fedotova E Y, Abramycheva N Y, Shabalina A A, Illarioshkin S N. Frontotemporal Dementia in Russia: Genetic Structure, Phenotypic Diversity, and Diagnostic Biomarkers. BCN 2025; 16 (1) :107-114
URL: http://bcn.iums.ac.ir/article-1-2774-en.html
URL: http://bcn.iums.ac.ir/article-1-2774-en.html
Yulia A. Shpilyukova *1 

 , Ekaterina Yu. Fedotova1 , Natalia Yu. Abramycheva1 , Alla A. Shabalina1 , Sergey N. Illarioshkin1
, Ekaterina Yu. Fedotova1 , Natalia Yu. Abramycheva1 , Alla A. Shabalina1 , Sergey N. Illarioshkin1
, Ekaterina Yu. Fedotova1 , Natalia Yu. Abramycheva1 , Alla A. Shabalina1 , Sergey N. Illarioshkin1
1- Research Center of Neurology, Moscow, Russia.
Keywords: Frontotemporal dementia (FTD), C9orf72, MAPT, GRN, Serum progranulin (PGRN), Amyloid β (Aβ-42), Phosphorylated tau protein (P-tau181)
Full-Text [PDF 890 kb]
| Abstract (HTML)
Full-Text:
1. Introduction
Frontotemporal dementia (FTD), a heterogeneous clinical syndrome corresponding to underlying frontotemporal lobar degeneration (FTLD), is one of the most common cause of dementia with early onset (Snowden et al., 2011). The annual incidence of FTLD in Europe is 2.36 cases per 100000 person-years, with the maximum peak of 13.09 cases per 100000 person-year at the age of 71 (Logroscino et al., 2023).
Typical clinical phenotypes of FTD include behavioral variant (bvFTD) (Rascovsky et al., 2011) and different variants of primary progressive aphasia: Non-fluent (nfvPPA), semantic (svPPA), and logopenic (lvPPA) (Gorno-Tempini et al., 2011). Cognitive, behavioral, and language symptoms are often accompanied by parkinsonism (Rowe, 2019) and amyotrophic lateral sclerosis (ALS) (Ng et al., 2015). In Europe, bvFTD represents the most common phenotype, followed by different variants of PPA, FTD-parkinsonism, and FTD-ALS (Logroscino et al., 2023). About 30% of FTD patients have a positive family history (Logroscino et al., 2023). The greatest contribution to the development of FTD is made by genes C9orf72, MAPT, and GRN, and the frequency of the corresponding mutations varies in different regions and populations (Moore et al., 2020).
FTD motor symptoms and speech phenotypes frequently overlap with other neurodegenerative diseases like Alzheimer disease (AD) and atypical parkinsonism syndromes (Alladi et al., 2007; Deutschländer et al., 2018). Behavioral symptoms of FTD are often challenging to differentiate from primary psychiatric diseases (Ducharme et al., 2020). These difficulties can delay FTD diagnosis by several years, so introducing genetic testing and other reliable biomarkers of this disease is very important (Ducharme et al., 2020).
Except for genetic testing, other biological biomarkers for FTD are absent. Among FTD patients, a progranulin glycoprotein (PGRN) level in biological fluids could be used to detect GRN mutation carriers (Antonell et al., 2012). Assessment of AD biomarkers in cerebrospinal fluid (CSF) in FTD is considered a potential option for more accurate excluding of AD pathology in FTD syndromes (Paraskevas et al., 2017; Casoli et al., 2019), but interpretation of these results could be complicated. On the one hand, in some pathological series, AD as the primary pathological diagnosis was established among 7.1% of patients with phenotype bvFTD, 44.1% with nfvPPA, 10% with svPPA, and 74.1% with mixed aphasia (Alladi et al., 2007). Nevertheless, among uncommon AD phenotypes, positivity on amyloid biomarkers could also be considered comorbid to another primary pathology (De Wilde et al., 2019; Naasan et al., 2016).
In the Russian population, there are no data on the incidence and prevalence of FTD, the frequency of common genetic variants, and the role of fluid biomarkers in diagnosis. Our study aimed to evaluate the phenotypic spectrum and genetic structure of FTD in a large cohort of Russian patients and assess the role of serum and CSF biomarkers that can be potentially used for the diagnosis.
2. Materials and Methods
Characteristics of the studied cohort
Our study used the local register of patients with FTD at the Department of Neurogenetics, Research Center of Neurology, Moscow. The database comprised clinical information on 226 patients with FTD: 126 women and 100 men, mean age 69±10 years (range 34–84), mean age of onset 62±10 years (range: 31–83). The phenotypical spectrum was presented by FTD (n=103), nfvPPA (n=46), svPPA (n=20), lvPPA (n=5), and undifferentiated forms of FTD and PPA (n=52). A positive family history was recorded for 65 patients (29%), and information on family history was unavailable for 47 patients (21%). All patients signed an informed, voluntary consent for the study.
Genetic testing
Genetic testing for the GGGGCC repeat expansion in the C9orf72 gene was performed in 193 patients. We used fragment analysis with repeated primed PCR, as described earlier (Lysogorskaia et al., 2016). The studied group was presented by bvFTD (n=85), nfvPPA (n=40), svPPA (n=14), lvPPA (n=5), and undifferentiated forms of FTD (n=49). The mean age of the disease onset was 61±9 years (range 35–80); 78 patients (40%) had positive family history.
Genetic testing for the GRN and MAPT genes point mutations was performed in 80 patients using the Sanger sequencing or massive parallel sequencing with our original “neurodegeneration” panel followed by Sanger sequencing confirmation. The group was presented by bvFTD (n=31), nfvPPA (n=22), svPPA (n=3), lvPPA (n=2), and undifferentiated forms of FTD (n=22). The mean age of the disease onset was 59±11 years (range 31–80); 32 patients had positive family history (40%). We also analyzed GRN and MAPT deletions and duplications in 50 patients (26 familial and 24 sporadic cases) by multiplex ligation-dependent probe amplification (MLPA) method (SALSA MLPA P275-C3, MRC-Holland, Netherlands) using the standard protocol. Polymerase chain reaction (PCR) products were separated by size with capillary electrophoresis using the genetic analyzer Nanofor 5 (Syntol, Russia). The analysis of the result was performed with GeneMarker software, version 3.0.1.
Serum PGRN level
Serum level of progranulin (PGRN), a product of the GRN gene, was assessed in 19 FTD patients, including 5 carriers of GRN mutations. According to the manufacturer protocol, we used enzyme-linked immunosorbent assay (ELISA) using reagents of Cloud Clone Corporation (USA, China). Since the level of PGRN can be influenced by various genetic factors, one of the most important is single nucleotide polymorphism (SNP) rs5848 in the GRN gene, which we assessed by Sanger sequencing.
AD biomarkers
We assessed AD biomarkers, amyloid-β(Aβ)-42 and phosphorylated tau protein (p-tau181), in CSF of 28 patients with FTD. The group was presented by bvFTD (n=16), nfvPPA (n=5), svPPA (n=3), and lvPPA (n=4). CSF biomarkers of Aβ-42 (cutoff point, 600 pg/mL) and p-tau181 (cutoff point, 50 pg/mL) were analyzed using ELISA according to the manufacturer’s protocol.
3. Results
C9orf72 gene
The pathological GGGGCC repeat expansion in the C9orf72 gene was found in 12 patients (6%), of which 8 had a positive family history. Thus, the frequency of C9orf72-associated FTD among familial cases was 10%, and among sporadic cases, 3.5%. The mean age of onset was 56.5±11 years (range 38–77). Men and women were represented equally. Clinical phenotype in most cases (7/12) was a combination of behavioral/aphatic FTD with ALS, while the other 5 cases were presented by “pure” bvFTD (n=3) and nfvPPA (n=2). Four patients also had parkinsonism with bradykinesia and muscle rigidity, and 5 had some other neurological signs, such as myoclonus, tremor (of the hands or head), and apraxia of swallowing. Apathy was the most common affective symptom (half of the cases), followed by depression, euphoria, disinhibition, and OCD. One patient with bvFTD and OCD also had frequent episodes of cognitive fluctuations with episodes of apathy and severe speech disturbances lasting up to several days (“Lewy body disease”-like phenotype).
GRN gene
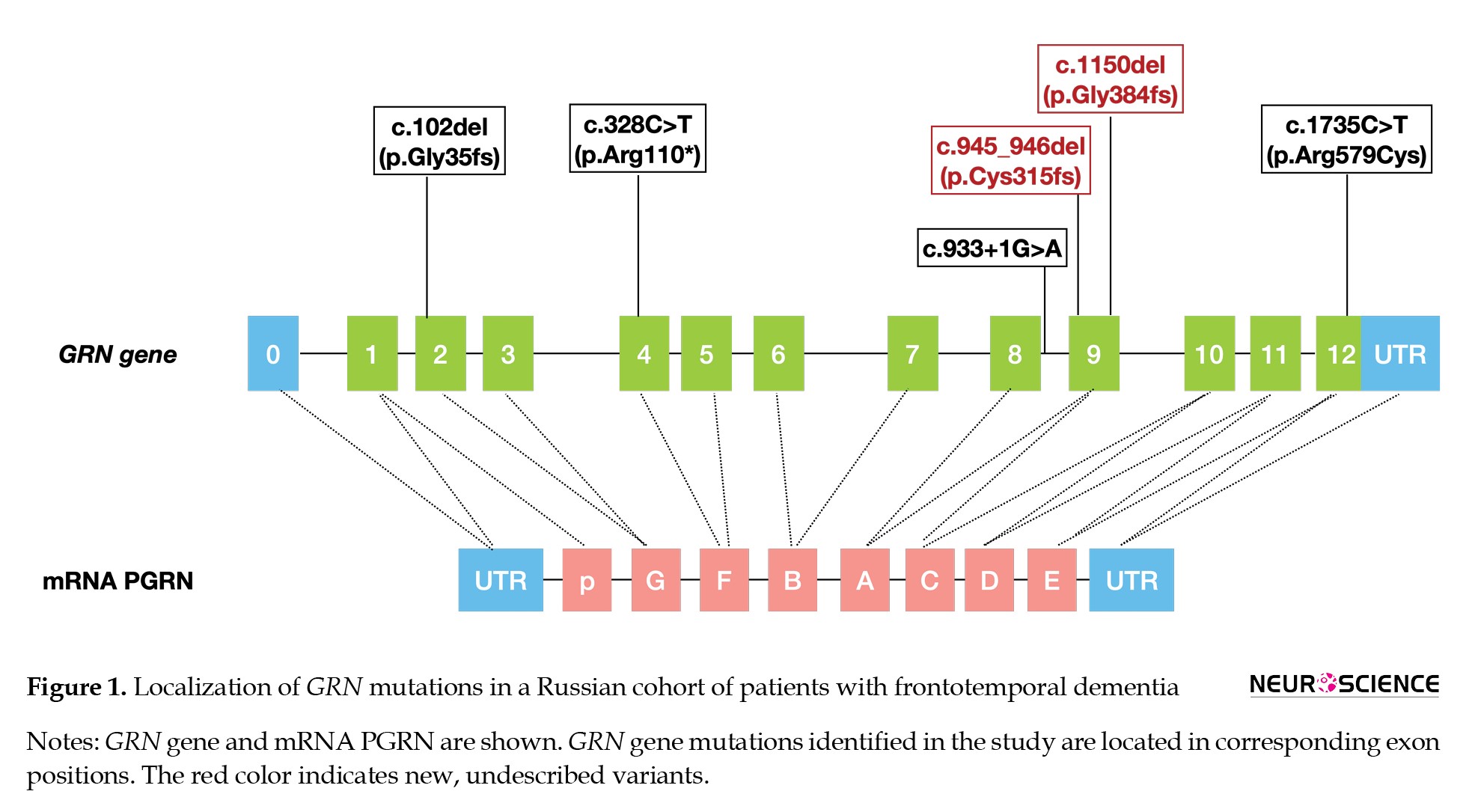
We identified 6 different GRN mutations in 10 unrelated patients (12.5% in our FTD cohort). Three variants were previously described as pathogenic: c.102del (p. Gly35fs) in exon 2 (4 patients), c.328C>T (p.Arg110*) in exon 4 (1 patient), and c.933+1G>A in the splice donor site (1 patient). We found 2 new frameshift deletions (both in exon 9) that could be interpreted as pathogenic: c.945_946del (p.Cys315fs) in 1 patient and c.1150delG (p.Gly384fs) in 2 patients. We also found a variant c.1735C>T (Arg579Cys) (rs748764855) in exon 12 with uncertain significance (VUS). This variant has low minor allele frequency (MAF) (T=0.000007, 1/140270, GnomAD; T=0.000025, 3/119780, ExAC) and was interpreted as “dangerous” with the SIFT program.
No deletions or duplications of GRN exons were found. All GRN mutations identified in our cohort are shown in Figure 1.
Frontotemporal dementia (FTD), a heterogeneous clinical syndrome corresponding to underlying frontotemporal lobar degeneration (FTLD), is one of the most common cause of dementia with early onset (Snowden et al., 2011). The annual incidence of FTLD in Europe is 2.36 cases per 100000 person-years, with the maximum peak of 13.09 cases per 100000 person-year at the age of 71 (Logroscino et al., 2023).
Typical clinical phenotypes of FTD include behavioral variant (bvFTD) (Rascovsky et al., 2011) and different variants of primary progressive aphasia: Non-fluent (nfvPPA), semantic (svPPA), and logopenic (lvPPA) (Gorno-Tempini et al., 2011). Cognitive, behavioral, and language symptoms are often accompanied by parkinsonism (Rowe, 2019) and amyotrophic lateral sclerosis (ALS) (Ng et al., 2015). In Europe, bvFTD represents the most common phenotype, followed by different variants of PPA, FTD-parkinsonism, and FTD-ALS (Logroscino et al., 2023). About 30% of FTD patients have a positive family history (Logroscino et al., 2023). The greatest contribution to the development of FTD is made by genes C9orf72, MAPT, and GRN, and the frequency of the corresponding mutations varies in different regions and populations (Moore et al., 2020).
FTD motor symptoms and speech phenotypes frequently overlap with other neurodegenerative diseases like Alzheimer disease (AD) and atypical parkinsonism syndromes (Alladi et al., 2007; Deutschländer et al., 2018). Behavioral symptoms of FTD are often challenging to differentiate from primary psychiatric diseases (Ducharme et al., 2020). These difficulties can delay FTD diagnosis by several years, so introducing genetic testing and other reliable biomarkers of this disease is very important (Ducharme et al., 2020).
Except for genetic testing, other biological biomarkers for FTD are absent. Among FTD patients, a progranulin glycoprotein (PGRN) level in biological fluids could be used to detect GRN mutation carriers (Antonell et al., 2012). Assessment of AD biomarkers in cerebrospinal fluid (CSF) in FTD is considered a potential option for more accurate excluding of AD pathology in FTD syndromes (Paraskevas et al., 2017; Casoli et al., 2019), but interpretation of these results could be complicated. On the one hand, in some pathological series, AD as the primary pathological diagnosis was established among 7.1% of patients with phenotype bvFTD, 44.1% with nfvPPA, 10% with svPPA, and 74.1% with mixed aphasia (Alladi et al., 2007). Nevertheless, among uncommon AD phenotypes, positivity on amyloid biomarkers could also be considered comorbid to another primary pathology (De Wilde et al., 2019; Naasan et al., 2016).
In the Russian population, there are no data on the incidence and prevalence of FTD, the frequency of common genetic variants, and the role of fluid biomarkers in diagnosis. Our study aimed to evaluate the phenotypic spectrum and genetic structure of FTD in a large cohort of Russian patients and assess the role of serum and CSF biomarkers that can be potentially used for the diagnosis.
2. Materials and Methods
Characteristics of the studied cohort
Our study used the local register of patients with FTD at the Department of Neurogenetics, Research Center of Neurology, Moscow. The database comprised clinical information on 226 patients with FTD: 126 women and 100 men, mean age 69±10 years (range 34–84), mean age of onset 62±10 years (range: 31–83). The phenotypical spectrum was presented by FTD (n=103), nfvPPA (n=46), svPPA (n=20), lvPPA (n=5), and undifferentiated forms of FTD and PPA (n=52). A positive family history was recorded for 65 patients (29%), and information on family history was unavailable for 47 patients (21%). All patients signed an informed, voluntary consent for the study.
Genetic testing
Genetic testing for the GGGGCC repeat expansion in the C9orf72 gene was performed in 193 patients. We used fragment analysis with repeated primed PCR, as described earlier (Lysogorskaia et al., 2016). The studied group was presented by bvFTD (n=85), nfvPPA (n=40), svPPA (n=14), lvPPA (n=5), and undifferentiated forms of FTD (n=49). The mean age of the disease onset was 61±9 years (range 35–80); 78 patients (40%) had positive family history.
Genetic testing for the GRN and MAPT genes point mutations was performed in 80 patients using the Sanger sequencing or massive parallel sequencing with our original “neurodegeneration” panel followed by Sanger sequencing confirmation. The group was presented by bvFTD (n=31), nfvPPA (n=22), svPPA (n=3), lvPPA (n=2), and undifferentiated forms of FTD (n=22). The mean age of the disease onset was 59±11 years (range 31–80); 32 patients had positive family history (40%). We also analyzed GRN and MAPT deletions and duplications in 50 patients (26 familial and 24 sporadic cases) by multiplex ligation-dependent probe amplification (MLPA) method (SALSA MLPA P275-C3, MRC-Holland, Netherlands) using the standard protocol. Polymerase chain reaction (PCR) products were separated by size with capillary electrophoresis using the genetic analyzer Nanofor 5 (Syntol, Russia). The analysis of the result was performed with GeneMarker software, version 3.0.1.
Serum PGRN level
Serum level of progranulin (PGRN), a product of the GRN gene, was assessed in 19 FTD patients, including 5 carriers of GRN mutations. According to the manufacturer protocol, we used enzyme-linked immunosorbent assay (ELISA) using reagents of Cloud Clone Corporation (USA, China). Since the level of PGRN can be influenced by various genetic factors, one of the most important is single nucleotide polymorphism (SNP) rs5848 in the GRN gene, which we assessed by Sanger sequencing.
AD biomarkers
We assessed AD biomarkers, amyloid-β(Aβ)-42 and phosphorylated tau protein (p-tau181), in CSF of 28 patients with FTD. The group was presented by bvFTD (n=16), nfvPPA (n=5), svPPA (n=3), and lvPPA (n=4). CSF biomarkers of Aβ-42 (cutoff point, 600 pg/mL) and p-tau181 (cutoff point, 50 pg/mL) were analyzed using ELISA according to the manufacturer’s protocol.
3. Results
C9orf72 gene
The pathological GGGGCC repeat expansion in the C9orf72 gene was found in 12 patients (6%), of which 8 had a positive family history. Thus, the frequency of C9orf72-associated FTD among familial cases was 10%, and among sporadic cases, 3.5%. The mean age of onset was 56.5±11 years (range 38–77). Men and women were represented equally. Clinical phenotype in most cases (7/12) was a combination of behavioral/aphatic FTD with ALS, while the other 5 cases were presented by “pure” bvFTD (n=3) and nfvPPA (n=2). Four patients also had parkinsonism with bradykinesia and muscle rigidity, and 5 had some other neurological signs, such as myoclonus, tremor (of the hands or head), and apraxia of swallowing. Apathy was the most common affective symptom (half of the cases), followed by depression, euphoria, disinhibition, and OCD. One patient with bvFTD and OCD also had frequent episodes of cognitive fluctuations with episodes of apathy and severe speech disturbances lasting up to several days (“Lewy body disease”-like phenotype).
GRN gene
We identified 6 different GRN mutations in 10 unrelated patients (12.5% in our FTD cohort). Three variants were previously described as pathogenic: c.102del (p. Gly35fs) in exon 2 (4 patients), c.328C>T (p.Arg110*) in exon 4 (1 patient), and c.933+1G>A in the splice donor site (1 patient). We found 2 new frameshift deletions (both in exon 9) that could be interpreted as pathogenic: c.945_946del (p.Cys315fs) in 1 patient and c.1150delG (p.Gly384fs) in 2 patients. We also found a variant c.1735C>T (Arg579Cys) (rs748764855) in exon 12 with uncertain significance (VUS). This variant has low minor allele frequency (MAF) (T=0.000007, 1/140270, GnomAD; T=0.000025, 3/119780, ExAC) and was interpreted as “dangerous” with the SIFT program.
No deletions or duplications of GRN exons were found. All GRN mutations identified in our cohort are shown in Figure 1.
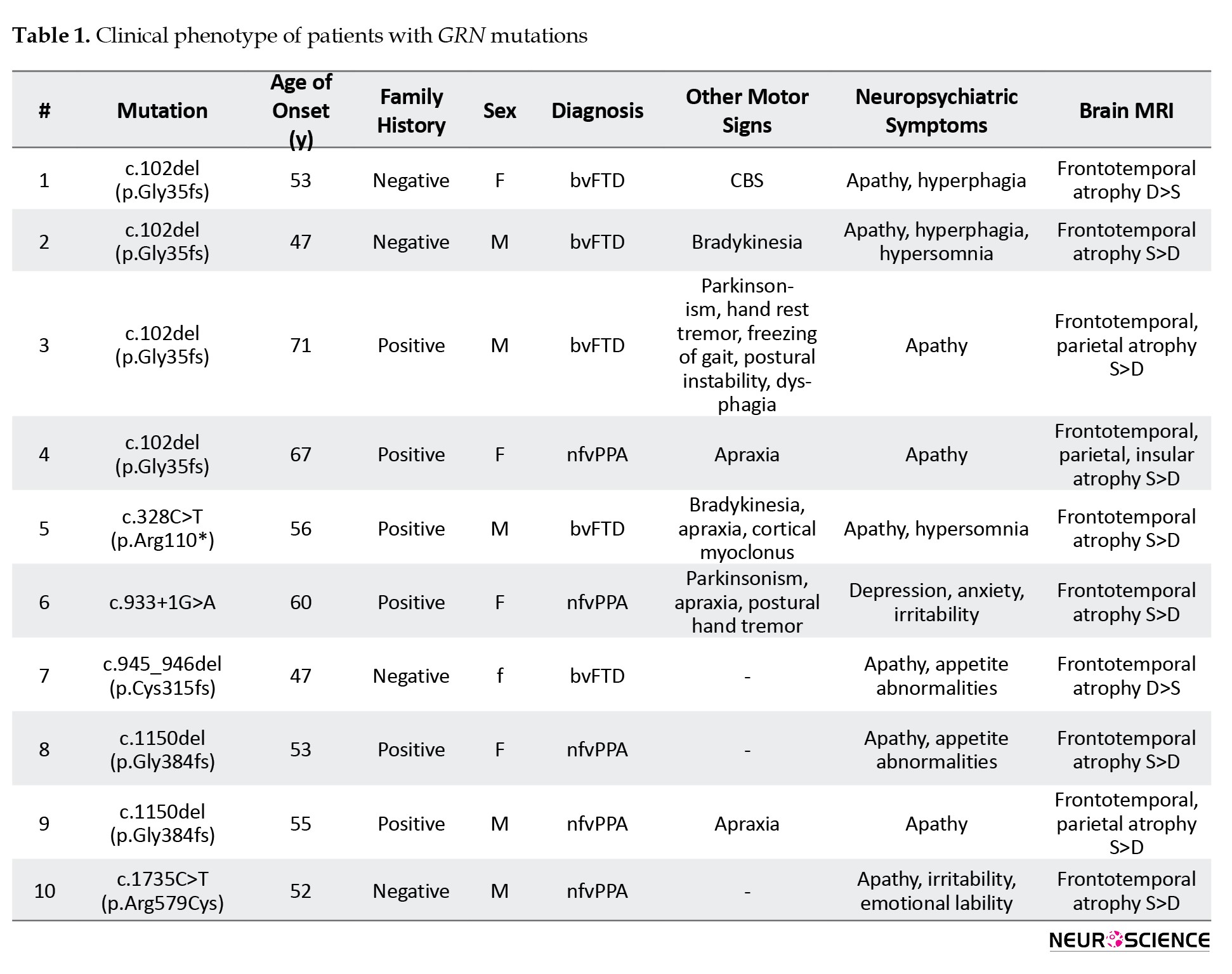
Six of 10 patients had a positive family history. Thus, the frequency of GRN-associated FTD among familial cases was 19%(6/32), and among sporadic cases, 8% (4/48). The mean age of onset was 57±8 years (47–71). Men and women were represented equally. The disease manifested in 5 cases with speech disorders (nfvPPA) and in 5with behavioral problems (bvFTD). Half of the patients showed signs of parkinsonism (one of them with corticobasal syndrome). In addition, the clinical picture of GRN-associated FTD cases included apraxia, postural hand tremor, myoclonus, postural instability, and dysphagia. Neuropsychiatric problems were presented mainly by apathy, appetite abnormalities, and hyperphagia; less often, one could see depression, anxiety, irritability, and hypersomnia. Detailed clinical phenotypes of patients with GRN mutations are presented in Table 1.
MAPT
We found 2 single nucleotide variants in the MAPT gene in our cohort of patients. One of them, c.1801C>G (p. Leu266Val) in exon 9, was previously described as pathogenic. A mutation carrier was a young male patient with a negative family history and a phenotype of primary progressive apraxia of speech with onset at 35 years. In addition to severe speech problems with fast progression to mutism, he had asymmetric apraxia and bradykinesia in the hands. Brain MRI showed asymmetric atrophy in the frontotemporal and parietal lobes with predominance on the right side.
The second variant, с.1505C>T (p.Ser502Phe), was found in exon 6 of the MAPT gene. This variant has low MAF (T=0.001604, 225/140278, GnomAD; T=0.001458, 177/121412 ExAC), and Clinvar database interpreted it as VUS. A carrier was a female patient with a positive family history and onset of bvFTD phenotype at the age of 74.
No deletions or duplications of MAPT exons were found. Thus, the frequency of MAPT mutations in the Russian cohort of FTD patients is low and represents 2.5%.
Serum PGRN
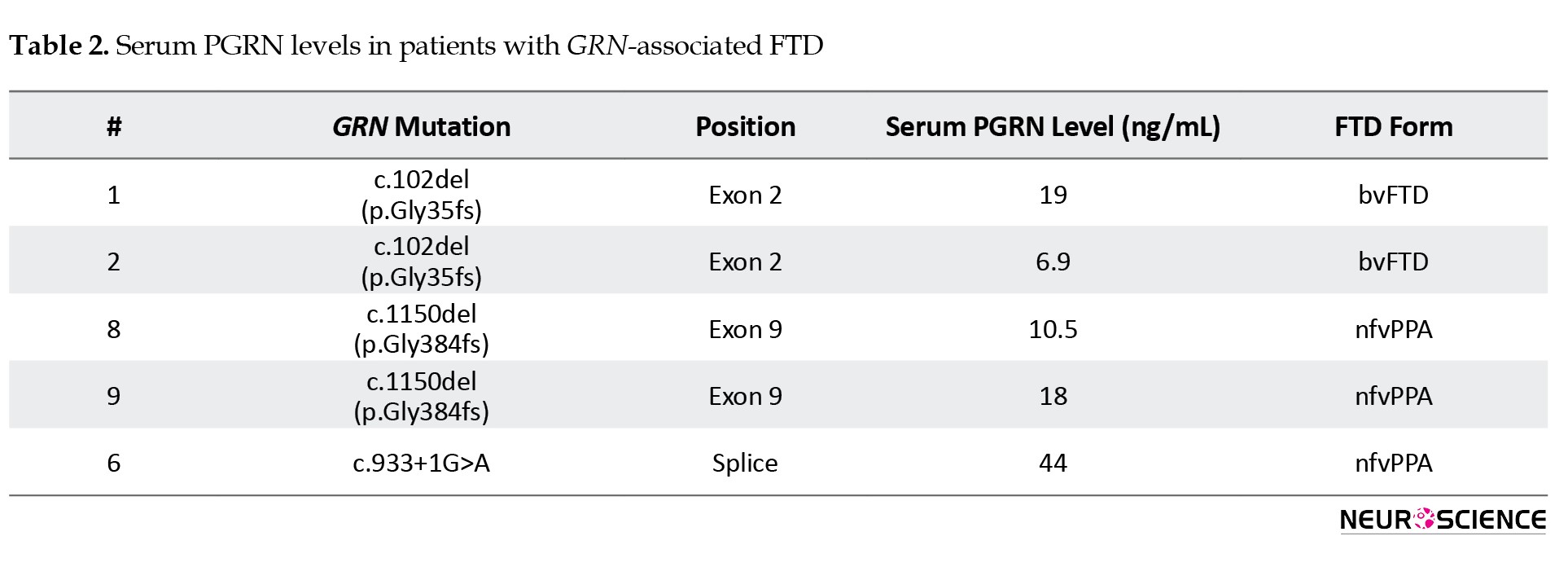
The serum PGRN levels in patients with exon mutations in the GRN gene were low compared to GRN-negative patients: 14 ng/mL [8;19] vs 31 [16; 45] ng/mL, respectively (P<0.05). Interestingly, patients with an intron GRN variant had a higher PGRN level than patients with exon mutations (Table 2).
We found no associations of PGRN levels with clinical, demographic factors, or rs5848 variants.
AD biomarkers
In most cases, we found normal CSF levels of Aβ-42 and p-tau181. However, 6 patients had decreased Aβ-42 levels and normal p-tau181 levels; among them, there were 3 patients with bvFTD and one patient each with svPPA, lvPPA, and nfvPPA.
4. Discussion
According to our data, the most common genetic form of FTD in the Russian cohort of patients is a GRN-associated form (12.5%), followed by forms with mutations in C9orf72 (6%) and MAPT (2.5%). The same distribution is characteristic for sporadic FTD (GRN – 8%, C9orf72 – 3.5%, and MAPT – 2%) and for familial cases of the disease (GRN – 19%, C9orf72 – 10% and MAPT – 3%). Among other populations, the closest data were obtained for the Italian, Portuguese, Belgian, and Scandinavian cohorts (Moore et al., 2020).
Our cohort of C9orf72-associated cases phenotypically presented by bvFTD, nfvPPA, and a combination of behavioral/aphatic FTD with ALS (most cases). The most common motor symptoms, except ALS-like, were parkinsonism and hyperkinetic disorders. The most common psychiatric features were apathy followed by depression, euphoria, disinhibition, and Obsessive compulsive disorder (OCD). It is important to know that patients with the GGGGCC repeat expansion in the C9orf72 gene usually have a high frequency of psychotic disorders, such as bipolar disorder, mania, major depressive episodes with catatonic features, and OCD (Ducharme et al., 2017). Those symptoms may precede typical FTD features by up to 4–5 years (Ducharme et al., 2017), which is a serious challenge for timely correct diagnosis. Thus, at the onset of FTD with affective disorders, the key to the correct diagnosis may be the identification of mild cognitive, speech, and motor disorders, which could serve as an indicator of the underlying FTLD. FTD patients carrying C9orf72 mutations have previously been shown to have a higher frequency of psychiatric symptoms and are less prone to eating behavior disorders and the loss of empathy compared to GRN mutation carriers (Snowden et al., 2015). Our results support these observations.
We found 6 mutations in the GRN gene, and 2 were not described previously. Mutation p.Gly35fs is the most frequent in our patients with typical phenotypes bvFTD or nfvPPA associated with parkinsonism. Another known mutation, p.Arg110*, was found in our patient with a usual phenotype of bvFTD associated with parkinsonism and cortical myoclonus. We have shown for the first time that a previously described variant c.933+1G>A can be characterized by an unusual phenotype, nfvPPA with parkinsonism, while Moore et al. (2020), in their meta-analysis described in carriers of this mutation only bvFTD, an AD-like phenotype or CBS. We also found two variants (p.Cys315fs and p.Gly384fs) not previously described in the literature, and the p.Gly384fs variant was found in two unrelated patients with low serum levels of PGRN.
Data on the serum PGRN level in patients with GRN-associated FTD remains controversial. PGRN level has previously been shown to be a reliable biomarker to predict GRN mutations even in an asymptomatic stage of neurodegeneration, and it does not correlate with the age of onset or clinical phenotype (Sellami et al., 2020). However, PGRN levels depend not only on GRN mutations but also on some SNPs (Hsiung, et al., 2011) and the presence of other diseases (Körtvélyessy et al., 2015; Vercellino et al., 2011; Yamamoto et al., 2014). In our study, serum PGRN levels decreased in patients with exon point mutations in GRN but not in patients with the intron c.933+1G>A variant. One can conclude that clarification of the diagnostic and biomarker role of the serum PGRN level requires further studies.
Our cohort identified only one previously described pathogenic mutation in the MAPT gene (Leu266Val). It is the first case of detection of a MAPT mutation in Russia. In another study, according to Moore et al. (2020), several patients with this mutation were observed with a mean age of onset of 32.4 years and a mean disease duration of 6 years. Most of the described patients had the phenotype of bvFTD; one had svPPA, and others had unspecified forms of dementia (Moore et al., 2020). Our patient had a typical age of onset but a unique clinical phenotype (fast progression of primary apraxia of speech and asymmetric apraxia/bradykinesia in the hands), which emphasizes the phenotypic diversity of a MAPT-associated form of the disease.
According to the literature, only 1.5% of individuals with GRN-associated forms have exon deletions or duplications in the GRN gene (Hsiung & Feldman, 2020), while there is no data about exon rearrangements in the MAPT gene. Our results indicate that this type of mutation is not typical for Russian patients with FTD.
We found changes in CSF biomarkers in 20% of studied cases (3/16 with bvFTD, 1/4 with lvPPA, 1/3 with svPPA, and 1/5 with nfvPPA). Our results are consistent with data on AD pathology prevalence and AD biomarkers positivity among patients with FTD-like phenotype (Alladi et al., 2017; Paraskevas et al., 2017). According to the current AD criteria, the diagnosis of AD is based on a combination of clinical phenotype and biomarker assessment (Dubois et al., 2021). lvPPA is a typical AD phenotype with a prevalent presentation of amyloid pathology in autopsy studies (Giannini et al., 2017), and its combination with Aβ-42 positivity could be regarded as 'probable' AD (Dubois et al., 2021). Interpreting uncommon AD phenotypes like behavioral, svPPA, and nfvPPA is more complicated because their combination with Aβ-42 positivity could be regarded as 'possible' AD according to IWG criteria (Dubois et al., 2021). Nevertheless, it is an unsolvable clinical situation because we do not yet have reliable markers of FTLD pathology, and in all these cases, we do not know whether it is its primary AD pathology or co-existing AD pathology. Some works discussed using a combination of two amyloid biomarkers (CSF and amyloid-PET) in unclear cases (Giacomucci et al., 2021).
5. Conclusion
It is the first data presenting the phenotypic spectrum, genetic structure, and biomarker assessment results in a large Russian cohort of FTD patients. Further studies may allow us to understand the prevalence of FTLD pathology in Russia and identify specific disease characteristics, facilitating early diagnosis. These data may also help to create algorithms for the differential diagnosis of FTD with other neurodegenerative dementias and, in the future, to find new therapeutic approaches and ways for the prevention of the disease in affected families.
Ethical Considerations
Compliance with ethical guidelines
This study was approved by the Local Ethics Committee of the Research Center of Neurology, Moscow, Russia (Code: 13-2/17).
Funding
The paper was partially extracted from the PhD dissertation Yulia A. Shpilyukova, approved by the Department of Neurogenetics, Research Center of Neurology, Moscow, Russia. The rest part of this research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
Conceptualization and supervision: Sergey Illarioshkin and Ekaterina Fedotova; Methodology: Ekaterina Fedotova and Yulia Shpilyukova; Data collection: Yulia Shpilyukova; Data analysis: Yulia Shpilyukova, Natalia Abramycheva and Alla Shabalina; Investigation, and writing the original draft: Yulia Shpilyukova; Review and editing: All authors.
Conflict of interest
The authors declared no conflict of interest.
Acknowledgments
The authors express their gratitude to the colleagues of the Research Center of Neurology and the I.M. Sechenov First Moscow State Medical University, Moscow, Russia for their assistance in collecting the material. Also, the authors express their gratitude to all patients who agreed to participate in the study.
References
Alladi, S., Xuereb, J., Bak, T., Nestor, P., Knibb, J., & Patterson, K., et al. (2007). Focal cortical presentations of Alzheimer's disease. Brain, 130(Pt 10), 2636–2645. [DOI:10.1093/brain/awm213] [PMID]
Antonell, A., Gil, S., Sánchez-Valle, R., Balasa, M., Bosch, B., & Prat, M. C., et al. (2012). Serum progranulin levels in patients with frontotemporal lobar degeneration and Alzheimer's disease: detection of GRN mutations in a Spanish cohort. Journal of Alzheimer's Disease, 31(3), 581–591. [DOI:10.3233/JAD-2012-112120] [PMID]
Casoli, T., Paolini, S., Fabbietti, P., Fattoretti, P., Paciaroni, L., & Fabi, K., et al. (2019). Cerebrospinal fluid biomarkers and cognitive status in differential diagnosis of frontotemporal dementia and Alzheimer's disease. The Journal of International Medical Research, 47(10), 4968–4980. [DOI:10.1177/0300060519860951] [PMID]
Deutschländer, A. B., Ross, O. A., Dickson, D. W., & Wszolek, Z. K. (2018). Atypical parkinsonian syndromes: A general neurologist's perspective. European Journal of Neurology, 25(1), 41–58. [DOI:10.1111/ene.13412] [PMID]
de Wilde, A., Reimand, J., Teunissen, C. E., Zwan, M., Windhorst, A. D., & Boellaard, R., et al. (2019). Discordant amyloid-β PET and CSF biomarkers and its clinical consequences. Alzheimer's Research & Therapy, 11(1), 78. [DOI:10.1186/s13195-019-0532-x] [PMID]
Dubois, B., Villain, N., Frisoni, G. B., Rabinovici, G. D., Sabbagh, M., & Cappa, S., et al. (2021). Clinical diagnosis of Alzheimer's disease: recommendations of the International Working Group. The Lancet. Neurology, 20(6), 484–496. [DOI:10.1016/S1474-4422(21)00066-1] [PMID]
Ducharme, S., Bajestan, S., Dickerson, B. C., & Voon, V. (2017). Psychiatric presentations of C9orf72 mutation: What are the diagnostic implications for clinicians? The Journal of Neuropsychiatry and Clinical Neurosciences, 29(3), 195–205.[DOI:10.1176/appi.neuropsych.16090168] [PMID]
Ducharme, S., Dols, A., Laforce, R., Devenney, E., Kumfor, F., & van den Stock, J., et al. (2020). Recommendations to distinguish behavioural variant frontotemporal dementia from psychiatric disorders. Brain, 143(6), 1632–1650. [DOI:10.1093/brain/awaa018] [PMID]
Giacomucci, G., Mazzeo, S., Bagnoli, S., Casini, M., Padiglioni, S., & Polito, C., et al. (2021). Matching clinical diagnosis and amyloid biomarkers in alzheimer's disease and frontotemporal dementia. Journal of Personalized Medicine, 11(1), 47. [DOI:10.3390/jpm11010047] [PMID]
Giannini, L. A., Irwin, D. J., McMillan, C. T., Ash, S., Rascovsky, K., Wolk, D. A., ... & Grossman, M. (2017). Clinical marker for Alzheimer disease pathology in logopenic primary progressive aphasia. Neurology, 88(24), 2276-2284. [DOI:10.1212/WNL.0000000000004034]
Gorno-Tempini, M. L., Hillis, A. E., Weintraub, S., Kertesz, A., Mendez, M., & Cappa, S. F., et al. (2011). Classification of primary progressive aphasia and its variants. Neurology, 76(11), 1006–1014. [DOI:10.1212/WNL.0b013e31821103e6] [PMID]
Hsiung, G. Y., Fok, A., Feldman, H. H., Rademakers, R., & Mackenzie, I. R. (2011). rs5848 polymorphism and serum progranulin level. Journal of the Neurological Sciences, 300(1-2), 28–32. [DOI:10.1016/j.jns.2010.10.009] [PMID]
Hsiung, G. R., & Feldman, H. H. (2020). GRN frontotemporal Dementia [Internet]. (2007). Retrieved from: [Link]
Körtvélyessy, P., Gukasjan, A., Sweeney-Reed, C. M., Heinze, H. J., Thurner, L., & Bittner, D. M. (2015). Progranulin and amyloid-β levels: Relationship to neuropsychology in frontotemporal and alzheimer's disease. Journal of Alzheimer's disease, 46(2), 375–380. [DOI:10.3233/JAD-150069] [PMID]
Logroscino, G., Piccininni, M., Graff, C., Hardiman, O., Ludolph, A. C., & Moreno, F., et al. (2023). Incidence of syndromes associated with frontotemporal lobar degeneration in 9 European countries. JAMA Neurology, 80(3), 279–286. [DOI:10.1001/jamaneurol.2022.5128] [PMID]
Lysogorskaia, E. V., Abramycheva, N. Y., Zakharova, M. N., Stepanova, M. S., Moroz, A. A., & Rossokhin, A. V., et al. (2015). Genetic studies of Russian patients with amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration, 17(1-2), 135–141.[DOI:10.3109/21678421.2015.1107100] [PMID]
Moore, K. M., Nicholas, J., Grossman, M., McMillan, C. T., Irwin, D. J., & Massimo, L., et al. (2020). Age at symptom onset and death and disease duration in genetic frontotemporal dementia: an international retrospective cohort study. The Lancet. Neurology, 19(2), 145–156. [DOI:10.1016/S1474-4422(19)30394-1] [PMID]
Naasan, G., Rabinovici, G. D., Ghosh, P., Elofson, J. D., Miller, B. L., & Coppola, G., et al. (2016). Amyloid in dementia associated with familial FTLD: not an innocent bystander. Neurocase, 22(1), 76–83. [PMID]
Ng, A. S., Rademakers, R., & Miller, B. L. (2015). Frontotemporal dementia: A bridge between dementia and neuromuscular disease. Annals of the New York Academy of Sciences, 1338(1), 71–93. [DOI:10.1111/nyas.12638] [PMID]
Paraskevas, G. P., Kasselimis, D., Kourtidou, E., Constantinides, V., Bougea, A., & Potagas, C., et al. (2017). Cerebrospinal fluid biomarkers as a diagnostic tool of the underlying pathology of primary progressive aphasia. Journal of Alzheimer's Disease, 55(4), 1453–1461. [DOI:10.3233/JAD-160494] [PMID]
Rascovsky, K., Hodges, J. R., Knopman, D., Mendez, M. F., Kramer, J. H., & Neuhaus, J., et al. (2011). Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain, 134(Pt 9), 2456–2477. [DOI:10.1093/brain/awr179] [PMID]
Rowe J. B. (2019). Parkinsonism in frontotemporal dementias. International Review of Neurobiology, 149, 249–275. [DOI:10.1016/bs.irn.2019.10.012] [PMID]
Sellami, L., Rucheton, B., Ben Younes, I., Camuzat, A., Saracino, D., & Rinaldi, D., et al. (2020). Plasma progranulin levels for frontotemporal dementia in clinical practice: A 10-year French experience. Neurobiology of Aging, 91, 167.e1–9. [DOI:10.1016/j.neurobiolaging.2020.02.014] [PMID]
Snowden, J. S., Adams, J., Harris, J., Thompson, J. C., Rollinson, S., & Richardson, A., et al. (2015). Distinct clinical and pathological phenotypes in frontotemporal dementia associated with MAPT, PGRN and C9orf72 mutations. Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration, 16(7-8), 497–505. [DOI:10.3109/21678421.2015.1074700] [PMID]
Snowden, J. S., Thompson, J. C., Stopford, C. L., Richardson, A. M., Gerhard, A., & Neary, D., et al. (2011). The clinical diagnosis of early-onset dementias: Diagnostic accuracy and clinicopathological relationships. Brain, 134(Pt 9), 2478–2492. [DOI:10.1093/brain/awr189] [PMID]
Vercellino, M., Grifoni, S., Romagnolo, A., Masera, S., Mattioda, A., & Trebini, C., et al. (2011). Progranulin expression in brain tissue and cerebrospinal fluid levels in multiple sclerosis. Multiple Sclerosis, 17(10), 1194–1201. [DOI:10.1177/1352458511406164] [PMID]
Yamamoto, Y., Takemura, M., Serrero, G., Hayashi, J., Yue, B., & Tsuboi, A., et al. (2014). Increased serum GP88 (Progranulin) concentrations in rheumatoid arthritis. Inflammation, 37(5), 1806–1813. [DOI:10.1007/s10753-014-9911-4] [PMID]
MAPT
We found 2 single nucleotide variants in the MAPT gene in our cohort of patients. One of them, c.1801C>G (p. Leu266Val) in exon 9, was previously described as pathogenic. A mutation carrier was a young male patient with a negative family history and a phenotype of primary progressive apraxia of speech with onset at 35 years. In addition to severe speech problems with fast progression to mutism, he had asymmetric apraxia and bradykinesia in the hands. Brain MRI showed asymmetric atrophy in the frontotemporal and parietal lobes with predominance on the right side.
The second variant, с.1505C>T (p.Ser502Phe), was found in exon 6 of the MAPT gene. This variant has low MAF (T=0.001604, 225/140278, GnomAD; T=0.001458, 177/121412 ExAC), and Clinvar database interpreted it as VUS. A carrier was a female patient with a positive family history and onset of bvFTD phenotype at the age of 74.
No deletions or duplications of MAPT exons were found. Thus, the frequency of MAPT mutations in the Russian cohort of FTD patients is low and represents 2.5%.
Serum PGRN
The serum PGRN levels in patients with exon mutations in the GRN gene were low compared to GRN-negative patients: 14 ng/mL [8;19] vs 31 [16; 45] ng/mL, respectively (P<0.05). Interestingly, patients with an intron GRN variant had a higher PGRN level than patients with exon mutations (Table 2).
We found no associations of PGRN levels with clinical, demographic factors, or rs5848 variants.
AD biomarkers
In most cases, we found normal CSF levels of Aβ-42 and p-tau181. However, 6 patients had decreased Aβ-42 levels and normal p-tau181 levels; among them, there were 3 patients with bvFTD and one patient each with svPPA, lvPPA, and nfvPPA.
4. Discussion
According to our data, the most common genetic form of FTD in the Russian cohort of patients is a GRN-associated form (12.5%), followed by forms with mutations in C9orf72 (6%) and MAPT (2.5%). The same distribution is characteristic for sporadic FTD (GRN – 8%, C9orf72 – 3.5%, and MAPT – 2%) and for familial cases of the disease (GRN – 19%, C9orf72 – 10% and MAPT – 3%). Among other populations, the closest data were obtained for the Italian, Portuguese, Belgian, and Scandinavian cohorts (Moore et al., 2020).
Our cohort of C9orf72-associated cases phenotypically presented by bvFTD, nfvPPA, and a combination of behavioral/aphatic FTD with ALS (most cases). The most common motor symptoms, except ALS-like, were parkinsonism and hyperkinetic disorders. The most common psychiatric features were apathy followed by depression, euphoria, disinhibition, and Obsessive compulsive disorder (OCD). It is important to know that patients with the GGGGCC repeat expansion in the C9orf72 gene usually have a high frequency of psychotic disorders, such as bipolar disorder, mania, major depressive episodes with catatonic features, and OCD (Ducharme et al., 2017). Those symptoms may precede typical FTD features by up to 4–5 years (Ducharme et al., 2017), which is a serious challenge for timely correct diagnosis. Thus, at the onset of FTD with affective disorders, the key to the correct diagnosis may be the identification of mild cognitive, speech, and motor disorders, which could serve as an indicator of the underlying FTLD. FTD patients carrying C9orf72 mutations have previously been shown to have a higher frequency of psychiatric symptoms and are less prone to eating behavior disorders and the loss of empathy compared to GRN mutation carriers (Snowden et al., 2015). Our results support these observations.
We found 6 mutations in the GRN gene, and 2 were not described previously. Mutation p.Gly35fs is the most frequent in our patients with typical phenotypes bvFTD or nfvPPA associated with parkinsonism. Another known mutation, p.Arg110*, was found in our patient with a usual phenotype of bvFTD associated with parkinsonism and cortical myoclonus. We have shown for the first time that a previously described variant c.933+1G>A can be characterized by an unusual phenotype, nfvPPA with parkinsonism, while Moore et al. (2020), in their meta-analysis described in carriers of this mutation only bvFTD, an AD-like phenotype or CBS. We also found two variants (p.Cys315fs and p.Gly384fs) not previously described in the literature, and the p.Gly384fs variant was found in two unrelated patients with low serum levels of PGRN.
Data on the serum PGRN level in patients with GRN-associated FTD remains controversial. PGRN level has previously been shown to be a reliable biomarker to predict GRN mutations even in an asymptomatic stage of neurodegeneration, and it does not correlate with the age of onset or clinical phenotype (Sellami et al., 2020). However, PGRN levels depend not only on GRN mutations but also on some SNPs (Hsiung, et al., 2011) and the presence of other diseases (Körtvélyessy et al., 2015; Vercellino et al., 2011; Yamamoto et al., 2014). In our study, serum PGRN levels decreased in patients with exon point mutations in GRN but not in patients with the intron c.933+1G>A variant. One can conclude that clarification of the diagnostic and biomarker role of the serum PGRN level requires further studies.
Our cohort identified only one previously described pathogenic mutation in the MAPT gene (Leu266Val). It is the first case of detection of a MAPT mutation in Russia. In another study, according to Moore et al. (2020), several patients with this mutation were observed with a mean age of onset of 32.4 years and a mean disease duration of 6 years. Most of the described patients had the phenotype of bvFTD; one had svPPA, and others had unspecified forms of dementia (Moore et al., 2020). Our patient had a typical age of onset but a unique clinical phenotype (fast progression of primary apraxia of speech and asymmetric apraxia/bradykinesia in the hands), which emphasizes the phenotypic diversity of a MAPT-associated form of the disease.
According to the literature, only 1.5% of individuals with GRN-associated forms have exon deletions or duplications in the GRN gene (Hsiung & Feldman, 2020), while there is no data about exon rearrangements in the MAPT gene. Our results indicate that this type of mutation is not typical for Russian patients with FTD.
We found changes in CSF biomarkers in 20% of studied cases (3/16 with bvFTD, 1/4 with lvPPA, 1/3 with svPPA, and 1/5 with nfvPPA). Our results are consistent with data on AD pathology prevalence and AD biomarkers positivity among patients with FTD-like phenotype (Alladi et al., 2017; Paraskevas et al., 2017). According to the current AD criteria, the diagnosis of AD is based on a combination of clinical phenotype and biomarker assessment (Dubois et al., 2021). lvPPA is a typical AD phenotype with a prevalent presentation of amyloid pathology in autopsy studies (Giannini et al., 2017), and its combination with Aβ-42 positivity could be regarded as 'probable' AD (Dubois et al., 2021). Interpreting uncommon AD phenotypes like behavioral, svPPA, and nfvPPA is more complicated because their combination with Aβ-42 positivity could be regarded as 'possible' AD according to IWG criteria (Dubois et al., 2021). Nevertheless, it is an unsolvable clinical situation because we do not yet have reliable markers of FTLD pathology, and in all these cases, we do not know whether it is its primary AD pathology or co-existing AD pathology. Some works discussed using a combination of two amyloid biomarkers (CSF and amyloid-PET) in unclear cases (Giacomucci et al., 2021).
5. Conclusion
It is the first data presenting the phenotypic spectrum, genetic structure, and biomarker assessment results in a large Russian cohort of FTD patients. Further studies may allow us to understand the prevalence of FTLD pathology in Russia and identify specific disease characteristics, facilitating early diagnosis. These data may also help to create algorithms for the differential diagnosis of FTD with other neurodegenerative dementias and, in the future, to find new therapeutic approaches and ways for the prevention of the disease in affected families.
Ethical Considerations
Compliance with ethical guidelines
This study was approved by the Local Ethics Committee of the Research Center of Neurology, Moscow, Russia (Code: 13-2/17).
Funding
The paper was partially extracted from the PhD dissertation Yulia A. Shpilyukova, approved by the Department of Neurogenetics, Research Center of Neurology, Moscow, Russia. The rest part of this research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
Conceptualization and supervision: Sergey Illarioshkin and Ekaterina Fedotova; Methodology: Ekaterina Fedotova and Yulia Shpilyukova; Data collection: Yulia Shpilyukova; Data analysis: Yulia Shpilyukova, Natalia Abramycheva and Alla Shabalina; Investigation, and writing the original draft: Yulia Shpilyukova; Review and editing: All authors.
Conflict of interest
The authors declared no conflict of interest.
Acknowledgments
The authors express their gratitude to the colleagues of the Research Center of Neurology and the I.M. Sechenov First Moscow State Medical University, Moscow, Russia for their assistance in collecting the material. Also, the authors express their gratitude to all patients who agreed to participate in the study.
References
Alladi, S., Xuereb, J., Bak, T., Nestor, P., Knibb, J., & Patterson, K., et al. (2007). Focal cortical presentations of Alzheimer's disease. Brain, 130(Pt 10), 2636–2645. [DOI:10.1093/brain/awm213] [PMID]
Antonell, A., Gil, S., Sánchez-Valle, R., Balasa, M., Bosch, B., & Prat, M. C., et al. (2012). Serum progranulin levels in patients with frontotemporal lobar degeneration and Alzheimer's disease: detection of GRN mutations in a Spanish cohort. Journal of Alzheimer's Disease, 31(3), 581–591. [DOI:10.3233/JAD-2012-112120] [PMID]
Casoli, T., Paolini, S., Fabbietti, P., Fattoretti, P., Paciaroni, L., & Fabi, K., et al. (2019). Cerebrospinal fluid biomarkers and cognitive status in differential diagnosis of frontotemporal dementia and Alzheimer's disease. The Journal of International Medical Research, 47(10), 4968–4980. [DOI:10.1177/0300060519860951] [PMID]
Deutschländer, A. B., Ross, O. A., Dickson, D. W., & Wszolek, Z. K. (2018). Atypical parkinsonian syndromes: A general neurologist's perspective. European Journal of Neurology, 25(1), 41–58. [DOI:10.1111/ene.13412] [PMID]
de Wilde, A., Reimand, J., Teunissen, C. E., Zwan, M., Windhorst, A. D., & Boellaard, R., et al. (2019). Discordant amyloid-β PET and CSF biomarkers and its clinical consequences. Alzheimer's Research & Therapy, 11(1), 78. [DOI:10.1186/s13195-019-0532-x] [PMID]
Dubois, B., Villain, N., Frisoni, G. B., Rabinovici, G. D., Sabbagh, M., & Cappa, S., et al. (2021). Clinical diagnosis of Alzheimer's disease: recommendations of the International Working Group. The Lancet. Neurology, 20(6), 484–496. [DOI:10.1016/S1474-4422(21)00066-1] [PMID]
Ducharme, S., Bajestan, S., Dickerson, B. C., & Voon, V. (2017). Psychiatric presentations of C9orf72 mutation: What are the diagnostic implications for clinicians? The Journal of Neuropsychiatry and Clinical Neurosciences, 29(3), 195–205.[DOI:10.1176/appi.neuropsych.16090168] [PMID]
Ducharme, S., Dols, A., Laforce, R., Devenney, E., Kumfor, F., & van den Stock, J., et al. (2020). Recommendations to distinguish behavioural variant frontotemporal dementia from psychiatric disorders. Brain, 143(6), 1632–1650. [DOI:10.1093/brain/awaa018] [PMID]
Giacomucci, G., Mazzeo, S., Bagnoli, S., Casini, M., Padiglioni, S., & Polito, C., et al. (2021). Matching clinical diagnosis and amyloid biomarkers in alzheimer's disease and frontotemporal dementia. Journal of Personalized Medicine, 11(1), 47. [DOI:10.3390/jpm11010047] [PMID]
Giannini, L. A., Irwin, D. J., McMillan, C. T., Ash, S., Rascovsky, K., Wolk, D. A., ... & Grossman, M. (2017). Clinical marker for Alzheimer disease pathology in logopenic primary progressive aphasia. Neurology, 88(24), 2276-2284. [DOI:10.1212/WNL.0000000000004034]
Gorno-Tempini, M. L., Hillis, A. E., Weintraub, S., Kertesz, A., Mendez, M., & Cappa, S. F., et al. (2011). Classification of primary progressive aphasia and its variants. Neurology, 76(11), 1006–1014. [DOI:10.1212/WNL.0b013e31821103e6] [PMID]
Hsiung, G. Y., Fok, A., Feldman, H. H., Rademakers, R., & Mackenzie, I. R. (2011). rs5848 polymorphism and serum progranulin level. Journal of the Neurological Sciences, 300(1-2), 28–32. [DOI:10.1016/j.jns.2010.10.009] [PMID]
Hsiung, G. R., & Feldman, H. H. (2020). GRN frontotemporal Dementia [Internet]. (2007). Retrieved from: [Link]
Körtvélyessy, P., Gukasjan, A., Sweeney-Reed, C. M., Heinze, H. J., Thurner, L., & Bittner, D. M. (2015). Progranulin and amyloid-β levels: Relationship to neuropsychology in frontotemporal and alzheimer's disease. Journal of Alzheimer's disease, 46(2), 375–380. [DOI:10.3233/JAD-150069] [PMID]
Logroscino, G., Piccininni, M., Graff, C., Hardiman, O., Ludolph, A. C., & Moreno, F., et al. (2023). Incidence of syndromes associated with frontotemporal lobar degeneration in 9 European countries. JAMA Neurology, 80(3), 279–286. [DOI:10.1001/jamaneurol.2022.5128] [PMID]
Lysogorskaia, E. V., Abramycheva, N. Y., Zakharova, M. N., Stepanova, M. S., Moroz, A. A., & Rossokhin, A. V., et al. (2015). Genetic studies of Russian patients with amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration, 17(1-2), 135–141.[DOI:10.3109/21678421.2015.1107100] [PMID]
Moore, K. M., Nicholas, J., Grossman, M., McMillan, C. T., Irwin, D. J., & Massimo, L., et al. (2020). Age at symptom onset and death and disease duration in genetic frontotemporal dementia: an international retrospective cohort study. The Lancet. Neurology, 19(2), 145–156. [DOI:10.1016/S1474-4422(19)30394-1] [PMID]
Naasan, G., Rabinovici, G. D., Ghosh, P., Elofson, J. D., Miller, B. L., & Coppola, G., et al. (2016). Amyloid in dementia associated with familial FTLD: not an innocent bystander. Neurocase, 22(1), 76–83. [PMID]
Ng, A. S., Rademakers, R., & Miller, B. L. (2015). Frontotemporal dementia: A bridge between dementia and neuromuscular disease. Annals of the New York Academy of Sciences, 1338(1), 71–93. [DOI:10.1111/nyas.12638] [PMID]
Paraskevas, G. P., Kasselimis, D., Kourtidou, E., Constantinides, V., Bougea, A., & Potagas, C., et al. (2017). Cerebrospinal fluid biomarkers as a diagnostic tool of the underlying pathology of primary progressive aphasia. Journal of Alzheimer's Disease, 55(4), 1453–1461. [DOI:10.3233/JAD-160494] [PMID]
Rascovsky, K., Hodges, J. R., Knopman, D., Mendez, M. F., Kramer, J. H., & Neuhaus, J., et al. (2011). Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain, 134(Pt 9), 2456–2477. [DOI:10.1093/brain/awr179] [PMID]
Rowe J. B. (2019). Parkinsonism in frontotemporal dementias. International Review of Neurobiology, 149, 249–275. [DOI:10.1016/bs.irn.2019.10.012] [PMID]
Sellami, L., Rucheton, B., Ben Younes, I., Camuzat, A., Saracino, D., & Rinaldi, D., et al. (2020). Plasma progranulin levels for frontotemporal dementia in clinical practice: A 10-year French experience. Neurobiology of Aging, 91, 167.e1–9. [DOI:10.1016/j.neurobiolaging.2020.02.014] [PMID]
Snowden, J. S., Adams, J., Harris, J., Thompson, J. C., Rollinson, S., & Richardson, A., et al. (2015). Distinct clinical and pathological phenotypes in frontotemporal dementia associated with MAPT, PGRN and C9orf72 mutations. Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration, 16(7-8), 497–505. [DOI:10.3109/21678421.2015.1074700] [PMID]
Snowden, J. S., Thompson, J. C., Stopford, C. L., Richardson, A. M., Gerhard, A., & Neary, D., et al. (2011). The clinical diagnosis of early-onset dementias: Diagnostic accuracy and clinicopathological relationships. Brain, 134(Pt 9), 2478–2492. [DOI:10.1093/brain/awr189] [PMID]
Vercellino, M., Grifoni, S., Romagnolo, A., Masera, S., Mattioda, A., & Trebini, C., et al. (2011). Progranulin expression in brain tissue and cerebrospinal fluid levels in multiple sclerosis. Multiple Sclerosis, 17(10), 1194–1201. [DOI:10.1177/1352458511406164] [PMID]
Yamamoto, Y., Takemura, M., Serrero, G., Hayashi, J., Yue, B., & Tsuboi, A., et al. (2014). Increased serum GP88 (Progranulin) concentrations in rheumatoid arthritis. Inflammation, 37(5), 1806–1813. [DOI:10.1007/s10753-014-9911-4] [PMID]
Type of Study: Original |
Subject:
Cellular and molecular Neuroscience
Received: 2023/08/10 | Accepted: 2024/07/29 | Published: 2025/01/1
Received: 2023/08/10 | Accepted: 2024/07/29 | Published: 2025/01/1
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
