

Volume 15, Issue 6 (November & December 2024)
BCN 2024, 15(6): 855-864 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Yuce Kahraman C, Kanjee M, Ercoskun P, Tatar A. Whole Exome Sequencing in Neurodevelopmental Disorders: A Single Center Study. BCN 2024; 15 (6) :855-864
URL: http://bcn.iums.ac.ir/article-1-2662-en.html
URL: http://bcn.iums.ac.ir/article-1-2662-en.html



1- Department of Medical Genetics, Faculty of Medicine, Ataturk University, Erzurum, Turkey.
Keywords: Neurodevelopmental disorder (NDD), Whole exome sequencing (WES), Developmental delay (DD) disorders, Intellectual disabilities, Congenital abnormalities
Full-Text [PDF 638 kb]
| Abstract (HTML)
Full-Text:
1. Introduction
Neurodevelopmental disorders (NDDs) are clinically and genetically a heterogeneous group of diseases. Epilepsy, intellectual disability (ID), autism spectrum disorder (ASD), and developmental delay (DD) are cataloged under NDDs (Stefanski et al., 2021).
The prevalence of DD and ID is reported to be 1%-3%. Congenital anomalies (CA) may accompany these features (Xiang et al., 2021). Genetic variants such as copy number variations (CNV), small insertions/deletions (indels), and single nucleotide variations (SNV) are responsible for these manifestations. The diagnostic yield of chromosomal microarray (CMA) for DD/ID and or CA and ASD is about 12%-28%. According to the 2010 American College of Medical Genetics and Genomics (ACMG) guidelines, CMA was the first-tier test for DD/ID and or CA (Manickam et al., 2021). With this test, SNV and indel could not be detected.
Over time, whole exome and genome sequencing (WES and or WGS) became widespread, and diagnostic rates of WES and or WGS for NDD were reported at 28%-68%. With the evaluation of these studies, the 2020 ACMG guideline recommended WES and or WGS for CA or DD/ID as the first-tier test. Thanks to technical additions, CNV analysis in WES is also possible (Malinowski et al., 2020).
It is difficult to diagnose this group of patients; constant visits to the hospital and undiagnosed returns cause emotional problems for the family. Identifying the underlying CA/DD/ID diagnosis may affect mortality and morbidity and reduce the burden on patients and families seeking answers (Malinowski et al., 2020). Finding the responsible gene will also be useful in understanding the pathogenesis and classifying this large group of patients. Due to the heterogeneous genetic background, WES analyses are more useful than single gene and panel tests in diagnosis (Stefanski et al., 2021). Treatment, prognosis, and follow-up processes of patients diagnosed with genetic tests are also improved, and therefore, a path can be followed according to the patient’s needs (Vickers & Gibson, 2019).
In this study, we aim to share the results of NDD patients who applied to our clinic and performed WES analysis and evaluate the rates of diagnosis with WES in our clinic.
2. Materials and Methods
Study patients
This study evaluated the pediatric patients admitted to our department with the diagnosis of NDD between March 2019 and May 2021 who underwent WES. Our study retrospectively evaluated the findings of 25 patients under 18 years old using medical genetics department records and the hospital automation system. Approval was obtained from our university’s Ethics Committee. Informed consent was taken from the parents of our patients.
Molecular study
According to the manufacturer’s protocols, genomic DNA was isolated from peripheral blood using a QIAamp DNA blood mini QIAcube kit (Qiagen, Hilden, Germany). All coding exons and exon-intron boundaries of the genes were amplified using the QIAseq human exome kit (Qiagen, Hilden, Germany). The prepared library was sequenced on the Illumina NextSeq platform (Illumina Inc., San Diego, CA, USA).
Bioinformatic analysis
The obtained data were analyzed using QIAGEN Clinical Insight (QCI) Interpret software (Qiagen, Hilden, Germany). More than 99% base-level coverage of targets at ≥20x was obtained. Sequencing data was aligned to the human reference genome, hg19. The QCI Interpret software includes the following underlying databases: Data reference sets and tools, QIAGEN Clinical Insight-Interpret (5.4.20190308), Ingenuity Knowledge Base (Stepford 190301.000), CADD (1.3), Allele Frequency Community (2018-09-06), EVS (ESP6500SI-V2), RefSeq Gene Model (2018-07-10), JASPAR (2013- 11), Ingenuity Knowledge Base Snapshot Timestamp (2019-03-01 11:17:42.0), Vista Enhancer hg19 (2012-07), PolyPhen (v2.2.2), 1000 Genome Frequency (phase3v5b), ExAC (0.3.1), iva (Oct 4 11:04 iva-1.0.736.jar), PhyloP hg19 (2009-11), DbSNP (151), GENCODE (release 28), CentoMD (5.0), OMIM (May 26, 2017), gnomAD (2.0.1), BSIFT (2016-02-23), TCGA (2013-09-05), Clinvar (2018-08-01), DGV (2016-05-15), HGMD (2018.3), and SIFT4G (2016-02-23). Variants were evaluated according to the ACMG criteria and classified as pathogenic, likely pathogenic, or variants of uncertain clinical significance.
Statistical analysis
The SPSS software, version 20 (IBM SPSS Statistics) was used for statistical analysis of the study. Descriptive statistics were used.
3. Results
The study comprises 14 male (56%) and 11(44%) female patients. Their Mean±SD age was 5.64±5.09 years (range: 1 to 16). DD in all 25 patients (100%), ID in 5(20%), epilepsy in 14 (56%), congenital anomaly (CA) in 10(40%), and other phenotypes such as autism, hypotonia, neuromotor regression in 3(12%) were seen.
CMA tests were requested before WES analysis in 8 patients (32%), and no results were obtained describing the phenotype. After WES analysis, we diagnosed 13 patients (52%) with pathogenic and likely pathogenic variants (Table 1), but 12(48%) remained unclear with variants of uncertain significance (VUS). However, we reclassified the variants as a result of segregation analysis.
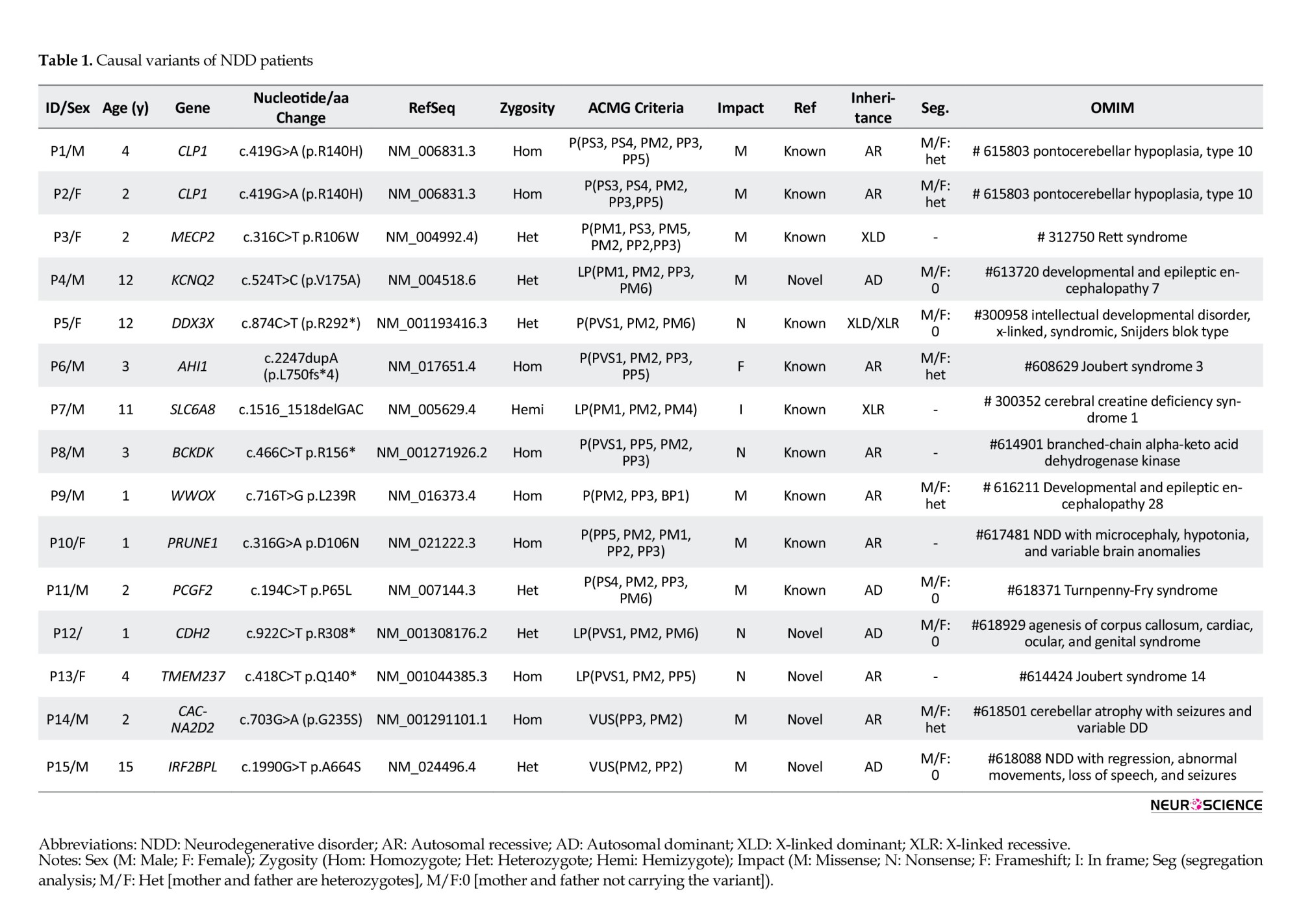
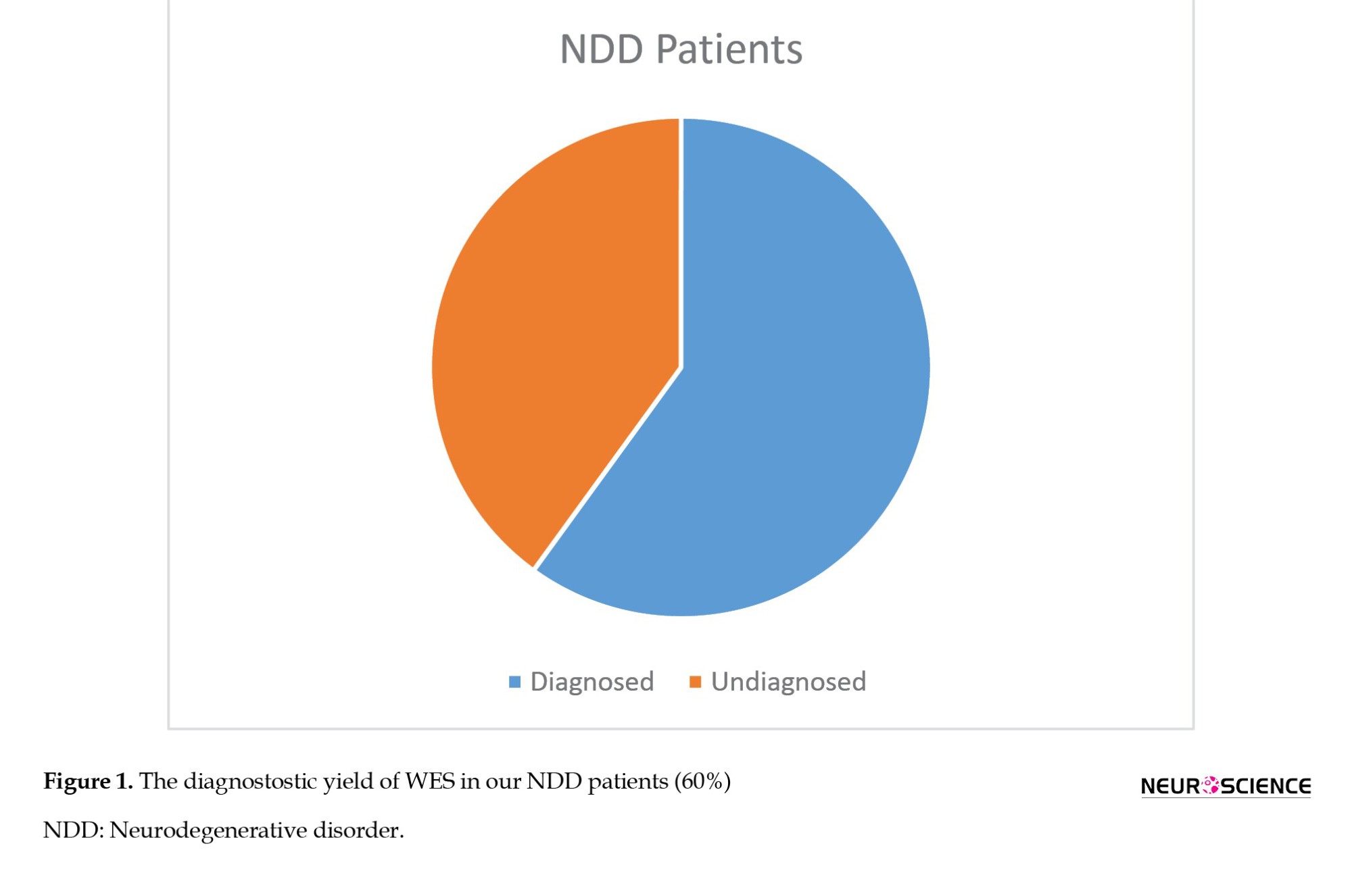
Thus, 2 patients that we previously classified as VUS were found to be phenotype-associated variants. We detected that the autosomal dominant (AD) VUS variant was de novo in one of the patients. In this case, we classified the variant as LP. The other patient also had a VUS associated with the phenotype. As a result of the segregation analysis, we determined that the homozygous variant was heterozygous in both parents and homozygous in a sibling who had a similar phenotype. We revised the variant as LP. Finally, the diagnostic yield of WES became 60% (Figure 1).
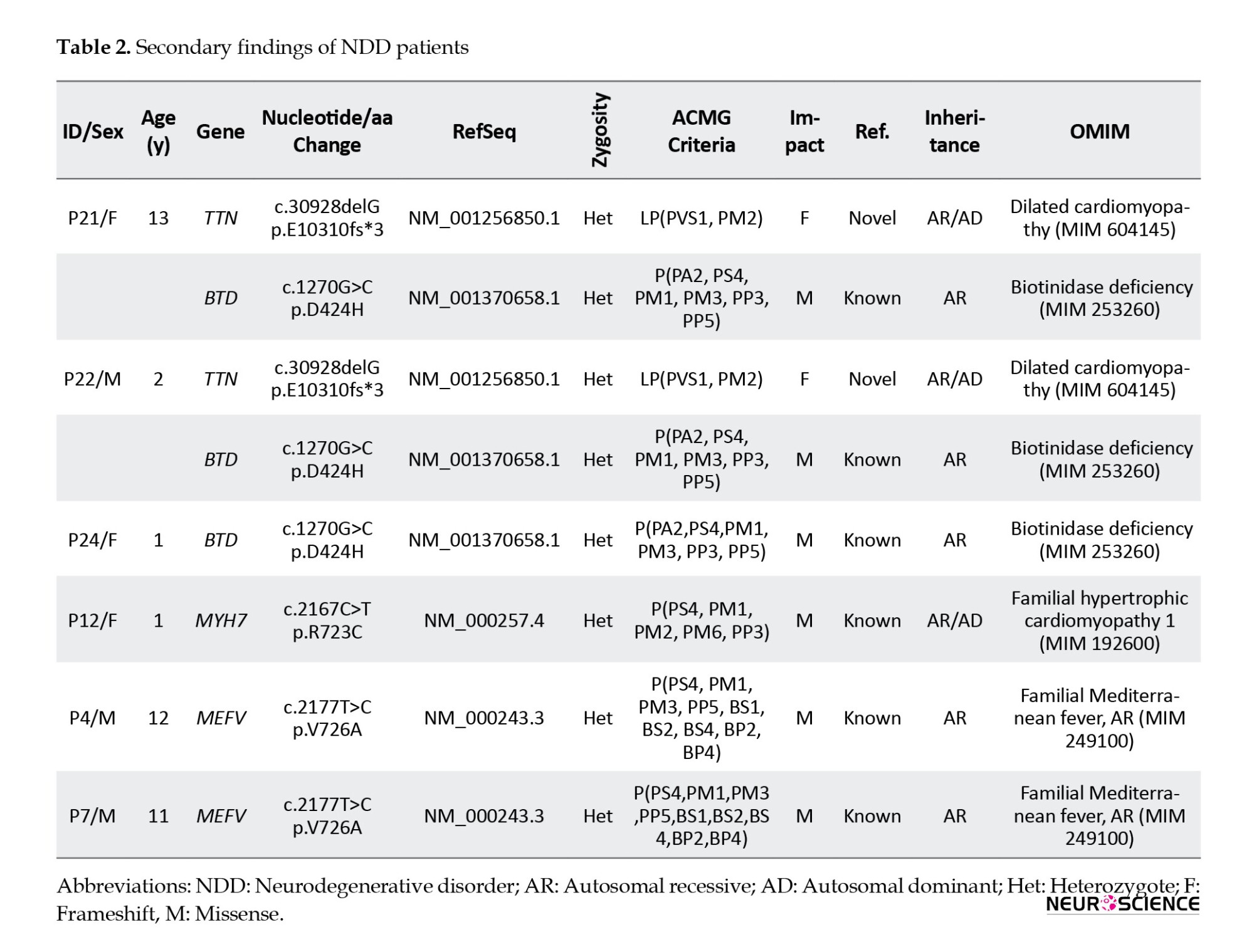
We included variants recommended to be reported as secondary findings in the ACMG guidelines in 4 patients. In addition, we included MEFV variants in the report of 2 patients due to the commonness of familial Mediterranean fever (FMF) in our country (Table 2)
4. Discussion
As in many clinically and genetically heterogeneous diseases, the rate of WES application is increasing in NDD. Many new responsible genes for NDD have been reported with WES analysis (Vissers et al., 2017; Xiang et al., 2021). Here, we evaluated the WES results of our 25 NDD patients and the diagnostic yield of WES in this group, which was 60%. This rate is in the upper range of the literature. In a meta-analysis, the diagnostic yield of clinical exome sequencing was reported as 36% overall, 31% for isolated NDD, and 53% for NDD plus associated conditions (Srivastava et al., 2019). Another meta-analysis study reported a diagnostic WES yield of 23.7% in NDD patients with epilepsy, ASD, or ID (Stefanski et al., 2021). A systematic evidence-based review reported a diagnostic yield of 28%-68% for WES and or WGS in NDD patients with DD/ID/CA (Malinowski et al., 2020).
In NDD patients with DD/ID/CA, the diagnostic yield of CMA has been reported to be 16%-28% (Malinowski et al., 2020). In our study, CMA analysis was performed on 32% of the patients, and none had a relevant result. Our WES analysis identified 14 variants of different genes that were responsible for the phenotypes. Five of these variants were novel, and the others were previously reported. Of the diagnosed diseases, 7 were autosomal recessive (AR), 4 were AD, and 3 were X-linked.
In two siblings, patient 1 and patient 2, a p.R140H pathogenic, homozygote, missense variant in the CLP1 gene was detected; AR pontocerebellar hypoplasia, type10 (OMIM: 615803) was diagnosed. p.R140H variant was reported as a founder mutation in Turkish families, and due to common consanguineous mating, its carrier frequency was 1/1000. It has been reported that this mutation disrupts tRNA splicing and causes progressive neurodegeneration (Schaffer et al., 2014). Global DD, lack of independent sitting or walking, seizures, lack of speech, and in MRI, pontocerebellar hypoplasia, cortical atrophy, and delayed myelinization were seen in the siblings consistent with the literature (Karaca et al., 2014; Schaffer et al., 2014).
In patient 3, the MECP2 p.R106W pathogenic variant was detected. The patient was diagnosed with Rett syndrome (OMIM: 312750). This syndrome is an X-linked dominant disease primarily seen in females with variable phenotypes due to X inactivation. Clinical findings begin with 6-18 months of age. Generally, there is no clinical manifestation in early infancy. Over time, head growth slows, and muscle tone decreases. Neuromotor delay and coordination disorder occur. Stereotypic hand movements, ataxic gait, scoliosis, constipation, excessive saliva, ID, periodic breathing, seizures, and verbal skill deterioration may occur (Sheikh et al., 2016). Our patient was a 2-year-old female presented with neuromotor delay, hypotonia, and seizures.
In patient 4, a novel p.V175A variant in the KCNQ2 gene was detected. The patient was diagnosed with developmental and epileptic encephalopathy 7, an AD disease. The segregation analysis showed no mutations in the parents, so the de novo variant was classified as likely pathogenic. It is an AD. NDD usually characterized by resistant seizures in the neonatal period, and de novo variants are responsible in most cases. Seizures usually resolve by 3 or 4 years of age, but neurological disorders are severe and persistent (Weckhuysen et al., 2012). The seizures of our patient started when he was a few months old and continued intermittently until he was 4 to 5 years old. He had spastic paraparesis and cortical atrophy in brain MRI, that is why his clinic was severe.
In patient 5, p.R292*, a pathogenic variant in the DDX3X gene was detected. DDX3X is responsible for syndromic X-linked mental retardation of the Snijders Blok type, which is predominantly seen in females. ID, microcephaly, movement disorders, behavioral problems such as ASD, hyperactivity, and epilepsy are seen in the disease (Snijders Blok et al., 2015). Our patient was a female with DD/ID, epilepsy, and ASD phenotype. Segregation analysis confirmed that the variant was de novo.
Patient 6 and patient 13 were diagnosed with Joubert syndrome 3 and 14, respectively. Both were inherited in an AR manner. Joubert syndrome is a group of diseases with genetic heterogeneity, characterized by symptoms such as neuroradiological ‘molar tooth sign,’ hypoplasia of the cerebellar vermis, irregularity of breathing pattern, and DD (Valente et al., 2005). p.L750fs*4 pathogenic, frameshift, and homozygote variant in the AHI1 gene were responsible for Joubert syndrome 3 in patient 6. It was confirmed that both parents were heterozygote carriers of the variant. Patient 6 had DD and the characteristic brain MRI findings. The novel, likely pathogenic p.Q140* variant in the TMEM237 gene was responsible for Joubert syndrome 14 in P13, who had DD, CA, hypotonia, and the characteristic brain MRI findings.
In patient 7, a likely pathogenic c.1516_1518delGAC variant in the SLC6A8 gene was detected as responsible for X-linked cerebral creatine deficiency syndrome. It is characterized by DD/ID, epilepsy, ASD, and severe speech delay (Salazar et al., 2020). Our patient had DD/ID, epilepsy, lack of speech, and microcephaly.
In patient 8, a pathogenic p.R156* homozygote variant in the BCKDK gene was detected that is responsible for branched-chain ketoacid dehydrogenase kinase deficiency. The same variant was reported in a Turkish family with ID, ASD, and epilepsy findings in 2012. In this study, clinical improvement was observed in mice with branched-chain amino acid supplementation. It was reported that patients may benefit from branched-chain amino acid supplementation (Novarino et al., 2012).
In patient 9, a pathogenic p.L239R homozygote variant in the WWOX gene was detected responsible for developmental and epileptic encephalopathy 28. It is an AR NDD in which resistant seizures, hypotonia, and psychomotor retardation are seen. Microcephaly, poor visual contact, and retinal degeneration may also be seen (Mignot et al., 2015). It was confirmed that both parents were heterozygote carriers of the variant. The patient had DD, epilepsy, and corpus callosum hypoplasia. His two older siblings are still living with tracheostomy; although they have not been tested yet, we suspected that they had the same diagnosis.
In patient 10, a pathogenic p.D106N homozygote variant in the PRUNE1 gene was detected responsible for NDD with microcephaly, hypotonia, and variable brain anomalies (OMIM: 617481). The same variant was reported in a Turkish family with microcephaly, cortical, and cerebellar atrophy (Karaca et al., 2015). Our patient had severe DD, refractory seizures, hypotonia, and cortical and cerebellar atrophy and died within a few months.
In patient 11, a pathogenic p. P65L heterozygote variant in the PCGF2 gene responsible for Turnpenny-Fry syndrome was detected. The segregation analysis showed that the variant was de novo, which was not found in the parents. It is an AD disorder with DD, ID, facial dysmorphism, and skeletal abnormalities (Ercoskun et al., 2021). Our patient had DD, facial dysmorphism, and neuromotor retardation.
In patient 12, a likely pathogenic novel p.R308* variant in the CDH2 gene was detected. It is responsible for agenesis of the corpus callosum, cardiac, ocular, and genital syndrome (OMIM:618929). The disorder is characterized by DD/ID, ocular, cardiac, and genital anomalies, corpus callosum hypoplasia, and craniofacial dysmorphisms (Accogli et al., 2019). Our patient had DD, corpus callosum hypoplasia, and nystagmus. The segregation analysis showed that the variant was de novo, which was not found in the parents.
In patient 14, a novel, homozygote p.G235S variant in the CACNA2D2 gene was detected. In silico tools predictions were as follows: CADD score, 27.4 (deleterious); PolyPhen, probably damaging; SIFT, damaging; PhyloP, not conserved; BLOSUM, MaxEntScan, B-SIFT, and QCI inferred activation, no prediction. According to ACMG criteria, the variant was classified as VUS. It was confirmed that both parents were heterozygote carriers of the variant. Besides, a similarly affected older sister had the same homozygote variant. We evaluated the variant as likely pathogenic with the segregation analysis data and consistent phenotype. CACNA2D2 is responsible for cerebellar atrophy with seizures and variable DD (OMIM:618501). It is an AR NDD characterized by cerebellar atrophy, severe refractory seizures in the first year of life, and DD (Butler et al., 2018). Our patient had seizures, cerebellar vermis atrophy, and DD, similar to the literature.
In patient 15, a novel, heterozygote p.A664S variant in the IRF2BPL gene was detected. In silico tools, predictions were as follows: CADD score,17.3 (likely deleterious); PolyPhen, benign; SIFT, tolerated, mutation taster; disease causing, PhyloP; not conserved, BLOSUM, MaxEntScan, GeneSplicer; B-SIFT, and QCI inferred activation; no prediction. According to ACMG criteria, the variant was classified as VUS. The segregation analysis showed that the variant was de novo, which was not found in the parents. We evaluated the variant as likely pathogenic with the segregation analysis data and consistent phenotype. IRF2BPL is responsible for NDDs with regression, abnormal movements, loss of speech, and seizures (OMIM:618088). In this disorder, psychomotor development is normal initially, followed by severe neurological regression and neurological findings (Tran Mau-Them et al., 2019). Our patient’s development was normal at the beginning. After the age of 9, the phenotype of a progressive gait, speech disorder, and repetitive movements emerged.
We also reported TTN, BTD, and MYH7 variants as the secondary findings for 4 patients that ACMG guidelines recommended reporting (Miller et al., 2021). Due to the prevalence of FMF in our country, we also reported the MEFV variants of 2 patients. Since all patients were heterozygous carriers for the mutations found, segregation analysis was not performed, but families were informed about the risk of carrying this mutation.
Since consanguineous marriage is common in our country, it is more likely to see different rare diseases compared to other countries. Our study also confirmed this information. Seven of the 15 diseases were inherited in an AR manner. Six parents were relatives; one was married and came from the same village.
Conclusion
Since it is possible to screen all coding exons associated with the phenotype in a single step, using the WES test is gradually increasing in diagnosing NDD diseases, considering its genetic heterogeneity. In our study, the diagnostic rate of NDD with WES was 60%. WES test, which is still expensive in our country, seems cost-effective when applied in well-selected patients compared to multiple single gene or panel tests. Determining the underlying cause of NDD will provide accurate diagnosis and clinical follow-up. It will even shed light on possible future gene therapy studies and ensure that families receive accurate genetic counseling.
Ethical Considerations
Compliance with ethical guidelines
This study was approved by the Clinical Research Ethic Committee of Ataturk University (Code: B.30.2.ATA.0.01.00/129). Written informed consent was taken from the parents of patients.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
Data collection: Cigdem Yuce Kahraman, Momen Kanjee, and Pelin Ercoskun; Conceptualization and study design, analysis, and writing: All authors.
Conflict of interest
The authors declared no conflict of interest.
References
Neurodevelopmental disorders (NDDs) are clinically and genetically a heterogeneous group of diseases. Epilepsy, intellectual disability (ID), autism spectrum disorder (ASD), and developmental delay (DD) are cataloged under NDDs (Stefanski et al., 2021).
The prevalence of DD and ID is reported to be 1%-3%. Congenital anomalies (CA) may accompany these features (Xiang et al., 2021). Genetic variants such as copy number variations (CNV), small insertions/deletions (indels), and single nucleotide variations (SNV) are responsible for these manifestations. The diagnostic yield of chromosomal microarray (CMA) for DD/ID and or CA and ASD is about 12%-28%. According to the 2010 American College of Medical Genetics and Genomics (ACMG) guidelines, CMA was the first-tier test for DD/ID and or CA (Manickam et al., 2021). With this test, SNV and indel could not be detected.
Over time, whole exome and genome sequencing (WES and or WGS) became widespread, and diagnostic rates of WES and or WGS for NDD were reported at 28%-68%. With the evaluation of these studies, the 2020 ACMG guideline recommended WES and or WGS for CA or DD/ID as the first-tier test. Thanks to technical additions, CNV analysis in WES is also possible (Malinowski et al., 2020).
It is difficult to diagnose this group of patients; constant visits to the hospital and undiagnosed returns cause emotional problems for the family. Identifying the underlying CA/DD/ID diagnosis may affect mortality and morbidity and reduce the burden on patients and families seeking answers (Malinowski et al., 2020). Finding the responsible gene will also be useful in understanding the pathogenesis and classifying this large group of patients. Due to the heterogeneous genetic background, WES analyses are more useful than single gene and panel tests in diagnosis (Stefanski et al., 2021). Treatment, prognosis, and follow-up processes of patients diagnosed with genetic tests are also improved, and therefore, a path can be followed according to the patient’s needs (Vickers & Gibson, 2019).
In this study, we aim to share the results of NDD patients who applied to our clinic and performed WES analysis and evaluate the rates of diagnosis with WES in our clinic.
2. Materials and Methods
Study patients
This study evaluated the pediatric patients admitted to our department with the diagnosis of NDD between March 2019 and May 2021 who underwent WES. Our study retrospectively evaluated the findings of 25 patients under 18 years old using medical genetics department records and the hospital automation system. Approval was obtained from our university’s Ethics Committee. Informed consent was taken from the parents of our patients.
Molecular study
According to the manufacturer’s protocols, genomic DNA was isolated from peripheral blood using a QIAamp DNA blood mini QIAcube kit (Qiagen, Hilden, Germany). All coding exons and exon-intron boundaries of the genes were amplified using the QIAseq human exome kit (Qiagen, Hilden, Germany). The prepared library was sequenced on the Illumina NextSeq platform (Illumina Inc., San Diego, CA, USA).
Bioinformatic analysis
The obtained data were analyzed using QIAGEN Clinical Insight (QCI) Interpret software (Qiagen, Hilden, Germany). More than 99% base-level coverage of targets at ≥20x was obtained. Sequencing data was aligned to the human reference genome, hg19. The QCI Interpret software includes the following underlying databases: Data reference sets and tools, QIAGEN Clinical Insight-Interpret (5.4.20190308), Ingenuity Knowledge Base (Stepford 190301.000), CADD (1.3), Allele Frequency Community (2018-09-06), EVS (ESP6500SI-V2), RefSeq Gene Model (2018-07-10), JASPAR (2013- 11), Ingenuity Knowledge Base Snapshot Timestamp (2019-03-01 11:17:42.0), Vista Enhancer hg19 (2012-07), PolyPhen (v2.2.2), 1000 Genome Frequency (phase3v5b), ExAC (0.3.1), iva (Oct 4 11:04 iva-1.0.736.jar), PhyloP hg19 (2009-11), DbSNP (151), GENCODE (release 28), CentoMD (5.0), OMIM (May 26, 2017), gnomAD (2.0.1), BSIFT (2016-02-23), TCGA (2013-09-05), Clinvar (2018-08-01), DGV (2016-05-15), HGMD (2018.3), and SIFT4G (2016-02-23). Variants were evaluated according to the ACMG criteria and classified as pathogenic, likely pathogenic, or variants of uncertain clinical significance.
Statistical analysis
The SPSS software, version 20 (IBM SPSS Statistics) was used for statistical analysis of the study. Descriptive statistics were used.
3. Results
The study comprises 14 male (56%) and 11(44%) female patients. Their Mean±SD age was 5.64±5.09 years (range: 1 to 16). DD in all 25 patients (100%), ID in 5(20%), epilepsy in 14 (56%), congenital anomaly (CA) in 10(40%), and other phenotypes such as autism, hypotonia, neuromotor regression in 3(12%) were seen.
CMA tests were requested before WES analysis in 8 patients (32%), and no results were obtained describing the phenotype. After WES analysis, we diagnosed 13 patients (52%) with pathogenic and likely pathogenic variants (Table 1), but 12(48%) remained unclear with variants of uncertain significance (VUS). However, we reclassified the variants as a result of segregation analysis.
Thus, 2 patients that we previously classified as VUS were found to be phenotype-associated variants. We detected that the autosomal dominant (AD) VUS variant was de novo in one of the patients. In this case, we classified the variant as LP. The other patient also had a VUS associated with the phenotype. As a result of the segregation analysis, we determined that the homozygous variant was heterozygous in both parents and homozygous in a sibling who had a similar phenotype. We revised the variant as LP. Finally, the diagnostic yield of WES became 60% (Figure 1).
We included variants recommended to be reported as secondary findings in the ACMG guidelines in 4 patients. In addition, we included MEFV variants in the report of 2 patients due to the commonness of familial Mediterranean fever (FMF) in our country (Table 2)
4. Discussion
As in many clinically and genetically heterogeneous diseases, the rate of WES application is increasing in NDD. Many new responsible genes for NDD have been reported with WES analysis (Vissers et al., 2017; Xiang et al., 2021). Here, we evaluated the WES results of our 25 NDD patients and the diagnostic yield of WES in this group, which was 60%. This rate is in the upper range of the literature. In a meta-analysis, the diagnostic yield of clinical exome sequencing was reported as 36% overall, 31% for isolated NDD, and 53% for NDD plus associated conditions (Srivastava et al., 2019). Another meta-analysis study reported a diagnostic WES yield of 23.7% in NDD patients with epilepsy, ASD, or ID (Stefanski et al., 2021). A systematic evidence-based review reported a diagnostic yield of 28%-68% for WES and or WGS in NDD patients with DD/ID/CA (Malinowski et al., 2020).
In NDD patients with DD/ID/CA, the diagnostic yield of CMA has been reported to be 16%-28% (Malinowski et al., 2020). In our study, CMA analysis was performed on 32% of the patients, and none had a relevant result. Our WES analysis identified 14 variants of different genes that were responsible for the phenotypes. Five of these variants were novel, and the others were previously reported. Of the diagnosed diseases, 7 were autosomal recessive (AR), 4 were AD, and 3 were X-linked.
In two siblings, patient 1 and patient 2, a p.R140H pathogenic, homozygote, missense variant in the CLP1 gene was detected; AR pontocerebellar hypoplasia, type10 (OMIM: 615803) was diagnosed. p.R140H variant was reported as a founder mutation in Turkish families, and due to common consanguineous mating, its carrier frequency was 1/1000. It has been reported that this mutation disrupts tRNA splicing and causes progressive neurodegeneration (Schaffer et al., 2014). Global DD, lack of independent sitting or walking, seizures, lack of speech, and in MRI, pontocerebellar hypoplasia, cortical atrophy, and delayed myelinization were seen in the siblings consistent with the literature (Karaca et al., 2014; Schaffer et al., 2014).
In patient 3, the MECP2 p.R106W pathogenic variant was detected. The patient was diagnosed with Rett syndrome (OMIM: 312750). This syndrome is an X-linked dominant disease primarily seen in females with variable phenotypes due to X inactivation. Clinical findings begin with 6-18 months of age. Generally, there is no clinical manifestation in early infancy. Over time, head growth slows, and muscle tone decreases. Neuromotor delay and coordination disorder occur. Stereotypic hand movements, ataxic gait, scoliosis, constipation, excessive saliva, ID, periodic breathing, seizures, and verbal skill deterioration may occur (Sheikh et al., 2016). Our patient was a 2-year-old female presented with neuromotor delay, hypotonia, and seizures.
In patient 4, a novel p.V175A variant in the KCNQ2 gene was detected. The patient was diagnosed with developmental and epileptic encephalopathy 7, an AD disease. The segregation analysis showed no mutations in the parents, so the de novo variant was classified as likely pathogenic. It is an AD. NDD usually characterized by resistant seizures in the neonatal period, and de novo variants are responsible in most cases. Seizures usually resolve by 3 or 4 years of age, but neurological disorders are severe and persistent (Weckhuysen et al., 2012). The seizures of our patient started when he was a few months old and continued intermittently until he was 4 to 5 years old. He had spastic paraparesis and cortical atrophy in brain MRI, that is why his clinic was severe.
In patient 5, p.R292*, a pathogenic variant in the DDX3X gene was detected. DDX3X is responsible for syndromic X-linked mental retardation of the Snijders Blok type, which is predominantly seen in females. ID, microcephaly, movement disorders, behavioral problems such as ASD, hyperactivity, and epilepsy are seen in the disease (Snijders Blok et al., 2015). Our patient was a female with DD/ID, epilepsy, and ASD phenotype. Segregation analysis confirmed that the variant was de novo.
Patient 6 and patient 13 were diagnosed with Joubert syndrome 3 and 14, respectively. Both were inherited in an AR manner. Joubert syndrome is a group of diseases with genetic heterogeneity, characterized by symptoms such as neuroradiological ‘molar tooth sign,’ hypoplasia of the cerebellar vermis, irregularity of breathing pattern, and DD (Valente et al., 2005). p.L750fs*4 pathogenic, frameshift, and homozygote variant in the AHI1 gene were responsible for Joubert syndrome 3 in patient 6. It was confirmed that both parents were heterozygote carriers of the variant. Patient 6 had DD and the characteristic brain MRI findings. The novel, likely pathogenic p.Q140* variant in the TMEM237 gene was responsible for Joubert syndrome 14 in P13, who had DD, CA, hypotonia, and the characteristic brain MRI findings.
In patient 7, a likely pathogenic c.1516_1518delGAC variant in the SLC6A8 gene was detected as responsible for X-linked cerebral creatine deficiency syndrome. It is characterized by DD/ID, epilepsy, ASD, and severe speech delay (Salazar et al., 2020). Our patient had DD/ID, epilepsy, lack of speech, and microcephaly.
In patient 8, a pathogenic p.R156* homozygote variant in the BCKDK gene was detected that is responsible for branched-chain ketoacid dehydrogenase kinase deficiency. The same variant was reported in a Turkish family with ID, ASD, and epilepsy findings in 2012. In this study, clinical improvement was observed in mice with branched-chain amino acid supplementation. It was reported that patients may benefit from branched-chain amino acid supplementation (Novarino et al., 2012).
In patient 9, a pathogenic p.L239R homozygote variant in the WWOX gene was detected responsible for developmental and epileptic encephalopathy 28. It is an AR NDD in which resistant seizures, hypotonia, and psychomotor retardation are seen. Microcephaly, poor visual contact, and retinal degeneration may also be seen (Mignot et al., 2015). It was confirmed that both parents were heterozygote carriers of the variant. The patient had DD, epilepsy, and corpus callosum hypoplasia. His two older siblings are still living with tracheostomy; although they have not been tested yet, we suspected that they had the same diagnosis.
In patient 10, a pathogenic p.D106N homozygote variant in the PRUNE1 gene was detected responsible for NDD with microcephaly, hypotonia, and variable brain anomalies (OMIM: 617481). The same variant was reported in a Turkish family with microcephaly, cortical, and cerebellar atrophy (Karaca et al., 2015). Our patient had severe DD, refractory seizures, hypotonia, and cortical and cerebellar atrophy and died within a few months.
In patient 11, a pathogenic p. P65L heterozygote variant in the PCGF2 gene responsible for Turnpenny-Fry syndrome was detected. The segregation analysis showed that the variant was de novo, which was not found in the parents. It is an AD disorder with DD, ID, facial dysmorphism, and skeletal abnormalities (Ercoskun et al., 2021). Our patient had DD, facial dysmorphism, and neuromotor retardation.
In patient 12, a likely pathogenic novel p.R308* variant in the CDH2 gene was detected. It is responsible for agenesis of the corpus callosum, cardiac, ocular, and genital syndrome (OMIM:618929). The disorder is characterized by DD/ID, ocular, cardiac, and genital anomalies, corpus callosum hypoplasia, and craniofacial dysmorphisms (Accogli et al., 2019). Our patient had DD, corpus callosum hypoplasia, and nystagmus. The segregation analysis showed that the variant was de novo, which was not found in the parents.
In patient 14, a novel, homozygote p.G235S variant in the CACNA2D2 gene was detected. In silico tools predictions were as follows: CADD score, 27.4 (deleterious); PolyPhen, probably damaging; SIFT, damaging; PhyloP, not conserved; BLOSUM, MaxEntScan, B-SIFT, and QCI inferred activation, no prediction. According to ACMG criteria, the variant was classified as VUS. It was confirmed that both parents were heterozygote carriers of the variant. Besides, a similarly affected older sister had the same homozygote variant. We evaluated the variant as likely pathogenic with the segregation analysis data and consistent phenotype. CACNA2D2 is responsible for cerebellar atrophy with seizures and variable DD (OMIM:618501). It is an AR NDD characterized by cerebellar atrophy, severe refractory seizures in the first year of life, and DD (Butler et al., 2018). Our patient had seizures, cerebellar vermis atrophy, and DD, similar to the literature.
In patient 15, a novel, heterozygote p.A664S variant in the IRF2BPL gene was detected. In silico tools, predictions were as follows: CADD score,17.3 (likely deleterious); PolyPhen, benign; SIFT, tolerated, mutation taster; disease causing, PhyloP; not conserved, BLOSUM, MaxEntScan, GeneSplicer; B-SIFT, and QCI inferred activation; no prediction. According to ACMG criteria, the variant was classified as VUS. The segregation analysis showed that the variant was de novo, which was not found in the parents. We evaluated the variant as likely pathogenic with the segregation analysis data and consistent phenotype. IRF2BPL is responsible for NDDs with regression, abnormal movements, loss of speech, and seizures (OMIM:618088). In this disorder, psychomotor development is normal initially, followed by severe neurological regression and neurological findings (Tran Mau-Them et al., 2019). Our patient’s development was normal at the beginning. After the age of 9, the phenotype of a progressive gait, speech disorder, and repetitive movements emerged.
We also reported TTN, BTD, and MYH7 variants as the secondary findings for 4 patients that ACMG guidelines recommended reporting (Miller et al., 2021). Due to the prevalence of FMF in our country, we also reported the MEFV variants of 2 patients. Since all patients were heterozygous carriers for the mutations found, segregation analysis was not performed, but families were informed about the risk of carrying this mutation.
Since consanguineous marriage is common in our country, it is more likely to see different rare diseases compared to other countries. Our study also confirmed this information. Seven of the 15 diseases were inherited in an AR manner. Six parents were relatives; one was married and came from the same village.
Conclusion
Since it is possible to screen all coding exons associated with the phenotype in a single step, using the WES test is gradually increasing in diagnosing NDD diseases, considering its genetic heterogeneity. In our study, the diagnostic rate of NDD with WES was 60%. WES test, which is still expensive in our country, seems cost-effective when applied in well-selected patients compared to multiple single gene or panel tests. Determining the underlying cause of NDD will provide accurate diagnosis and clinical follow-up. It will even shed light on possible future gene therapy studies and ensure that families receive accurate genetic counseling.
Ethical Considerations
Compliance with ethical guidelines
This study was approved by the Clinical Research Ethic Committee of Ataturk University (Code: B.30.2.ATA.0.01.00/129). Written informed consent was taken from the parents of patients.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
Data collection: Cigdem Yuce Kahraman, Momen Kanjee, and Pelin Ercoskun; Conceptualization and study design, analysis, and writing: All authors.
Conflict of interest
The authors declared no conflict of interest.
References
Accogli, A., Calabretta, S., St-Onge, J., Boudrahem-Addour, N., Dionne-Laporte, A., & Joset, P., et al. (2019). De Novo pathogenic variants in N-cadherin cause a syndromic neurodevelopmental disorder with corpus collosum, axon, cardiac, ocular, and genital defects. American Journal of Human Genetics, 105(4), 854–868. [PMID]
Butler, K. M., Holt, P. J., Milla, S. S., da Silva, C., Alexander, J. J., & Escayg, A. (2018). Epileptic encephalopathy and cerebellar atrophy resulting from compound heterozygous Cacna2d2 Variants. Case Reports in Genetics, 2018, 6308283.[DOI:10.1155/2018/6308283] [PMID]
Ercoskun, P., Yuce Kahraman, C., Adanur Saglam, K., Kanjee, M., & Tatar, A. (2022). A new case of turnpenny-fry syndrome. American Journal of Medical Genetics. Part A, 188(2), 688–691. [DOI:10.1002/ajmg.a.62560] [PMID]
Karaca, E., Harel, T., Pehlivan, D., Jhangiani, S. N., Gambin, T., & Coban Akdemir, Z., et al. (2015). Genes that affect brain structure and function identified by rare variant analyses of mendelian neurologic disease. Neuron, 88(3), 499–513.[DOI:10.1016/j.neuron.2015.09.048] [PMID]
Karaca, E., Weitzer, S., Pehlivan, D., Shiraishi, H., Gogakos, T., & Hanada, T., et al. (2014). Human Clp1 mutations alter trna biogenesis, affecting both peripheral and central nervous system function. Cell, 157(3), 636–650. [DOI:10.1016/j.cell.2014.02.058] [PMID]
Malinowski, J., Miller, D. T., Demmer, L., Gannon, J., Pereira, E. M., & Schroeder, M. C., et al. (2020). Systematic evidence-based review: Outcomes from exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 22(6), 986–1004. [DOI:10.1038/s41436-020-0771-z] [PMID]
Manickam, K., McClain, M. R., Demmer, L. A., Biswas, S., Kearney, H. M., & Malinowski, J., et al. (2021). Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: An evidence-based clinical guideline of the American College Of Medical Genetics And Genomics (ACMG). Genetics in Medicine: Official Journal of The American College of Medical Genetics, 23(11), 2029–2037. [DOI:10.1038/s41436-021-01242-6] [PMID]
Mignot, C., Lambert, L., Pasquier, L., Bienvenu, T., Delahaye-Duriez, A., & Keren, B., et al. (2015). WWox-related encephalopathies: Delineation of the phenotypical spectrum and emerging genotype-phenotype correlation. Journal of Medical Genetics, 52(1), 61–70. [DOI:10.1136/jmedgenet-2014-102748] [PMID]
Miller, D. T., Lee, K., Gordon, A. S., Amendola, L. M., Adelman, K., & Bale, S. J., K., et al. (2021). Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2021 update: A policy statement of the American College Of Medical Genetics And Genomics (Acmg). Genetics in medicine: Official Journal of the American College of Medical Genetics, 23(8), 1391–1398. [DOI:10.1038/s41436-021-01171-4] [PMID]
Novarino, G., El-Fishawy, P., Kayserili, H., Meguid, N. A., Scott, E. M., & Schroth, J., et al. (2012). Mutations in bckd-kinase lead to a potentially treatable form of autism with epilepsy. Science (New York, N.Y.), 338(6105), 394–397. [DOI:10.1126/science.1224631] [PMID]
Salazar, M. D., Zelt, N. B., Saldivar, R., Kuntz, C. P., Chen, S., & Penn, W. D., et al. (2020). Classification of the molecular defects associated with pathogenic variants of the slc6a8 creatine transporter. Biochemistry, 59(13), 1367–1377. [DOI:10.1021/acs.biochem.9b00956] [PMID]
Schaffer, A. E., Eggens, V. R., Caglayan, A. O., Reuter, M. S., Scott, E., & Coufal, N. G., et al. (2014). Clp1 founder mutation links trna splicing and maturation to cerebellar development and neurodegeneration. Cell, 157(3), 651–663. [DOI:10.1016/j.cell.2014.03.049] [PMID]
Sheikh, T. I., Ausió, J., Faghfoury, H., Silver, J., Lane, J. B., & Eubanks, J. H., et al. (2016). From function to phenotype: Impaired Dna binding and clustering correlates with clinical severity in males with missense mutations in MECP2. Scientific Reports, 6, 38590. [DOI:10.1038/srep38590] [PMID]
Snijders Blok, L., Madsen, E., Juusola, J., Gilissen, C., Baralle, D., & Reijnders, M. R., et al. (2015). Mutations In Ddx3x are a common cause of unexplained intellectual disability with gender-specific effects on wnt signaling. American Journal of Human Genetics, 97(2), 343–352. [DOI:10.1016/j.ajhg.2015.07.004] [PMID]
Srivastava, S., Love-Nichols, J. A., Dies, K. A., Ledbetter, D. H., Martin, C. L., & Chung, W. K., et al. (2019). Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genetics in Medicine: Official Journal of The American College of Medical Genetics, 21(11), 2413–2421. [DOI:10.1038/s41436-019-0554-6] [PMID]
Stefanski, A., Calle-López, Y., Leu, C., Pérez-Palma, E., Pestana-Knight, E., & Lal, D. (2021). Clinical sequencing yield in epilepsy, autism spectrum disorder, and intellectual disability: A systematic review and meta-analysis. Epilepsia, 62(1), 143-151. [DOI:10.1111/epi.16755] [PMID]
Tran Mau-Them, F., Guibaud, L., Duplomb, L., Keren, B., Lindstrom, K., & Marey, I., et al. (2019). De Novo truncating variants in the intronless Irf2bpl are responsible for developmental epileptic encephalopathy. Genetics in Medicine, 21(4), 1008–1014. [DOI:10.1038/s41436-018-0143-0] [PMID]
Valente, E. M., Marsh, S. E., Castori, M., Dixon-Salazar, T., Bertini, E., & Al-Gazali, L., et al. (2005). Distinguishing the four genetic causes of jouberts syndrome-related disorders. Annals of Neurology, 57(4), 513–519. [DOI:10.1002/ana.20422] [PMID]
Vickers, R. R., & Gibson, J. S. (2019). A review of the genomic analysis of children presenting with developmental delay/intellectual disability and associated dysmorphic features. Cureus, 11(1), e3873. [DOI:10.7759/cureus.3873] [PMID]
Vissers, L. E. L. M., van Nimwegen, K. J. M., Schieving, J. H., Kamsteeg, E. J., Kleefstra, T., & Yntema, H. G., et al. (2017). A clinical utility study of exome sequencing versus conventional genetic testing in pediatric neurology. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 19(9), 1055–1063. [DOI:10.1038/gim.2017.1] [PMID]
Type of Study: Original |
Subject:
Cellular and molecular Neuroscience
Received: 2023/02/1 | Accepted: 2024/02/10 | Published: 2024/11/1
Received: 2023/02/1 | Accepted: 2024/02/10 | Published: 2024/11/1
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
