

Volume 15, Issue 3 (May & Jun 2024)
BCN 2024, 15(3): 333-342 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Parvishan A, Taghi Joghataei M, Kiani J, Shahbazi A, Faghihi F, Ghadiri M. Generation of Human-induced Pluripotent Stem Cells Derived From Dermal Fibroblast of Schizophrenic Patients. BCN 2024; 15 (3) :333-342
URL: http://bcn.iums.ac.ir/article-1-2282-en.html
URL: http://bcn.iums.ac.ir/article-1-2282-en.html
Asghar Parvishan1 

 , Mohammad Taghi Joghataei *2 , Jafar Kiani3 , Ali Shahbazi1 , Faezeh Faghihi2 , Mohammad Ghadiri4
, Mohammad Taghi Joghataei *2 , Jafar Kiani3 , Ali Shahbazi1 , Faezeh Faghihi2 , Mohammad Ghadiri4
, Mohammad Taghi Joghataei *2 , Jafar Kiani3 , Ali Shahbazi1 , Faezeh Faghihi2 , Mohammad Ghadiri4
1- Department of Neuroscience, Faculty of Advanced Technologies in Medicine, Iran University of Medical Sciences, Tehran, Iran.
2- Cellular and Molecular Research Center, Iran University of Medical Sciences, Tehran, Iran.
3- Department of Molecular Medicine, Faculty of Advanced Technologies in Medicine, Iran University of Medical Sciences, Tehran, Iran.
4- Psychiatry Center of Iran, Iran University of Medical Sciences, Tehran, Iran.
2- Cellular and Molecular Research Center, Iran University of Medical Sciences, Tehran, Iran.
3- Department of Molecular Medicine, Faculty of Advanced Technologies in Medicine, Iran University of Medical Sciences, Tehran, Iran.
4- Psychiatry Center of Iran, Iran University of Medical Sciences, Tehran, Iran.
Full-Text [PDF 2471 kb]
| Abstract (HTML)
Full-Text:
1. Introduction
Schizophrenia (SCZ) is a severe neuropsychiatric disorder with a global prevalence of 1% and 80-85% heritability (Sullivan et al., 2003). Multiple reasons, including genetic and environmental factors, have roles in the pathogenesis of SCZ (Tsuang et al., 2001; Palha & Goodman, 2006), and perturbation of these agents could cause the disturbance of information of the neural network and brain function (Christian et al., 2010). SCZ onset often happens in early adulthood (Sawa & Snyder, 2002), but in some patients, the abnormal neurodevelopmental process may start in childhood (Weinberger, 1987; Marin, 2012). Furthermore, most patients show different responses to treatment (Schennach et al., 2012), and only a few percent effectively respond to the treatment (Emsley et al., 2011). Studies have shown that SCZ is accompanied by abnormal neuronal communication with a significant role for oligodendrocyte and progenitor cells in the pathogenesis (Rubinov, 2013; Federspiel et al., 2006; Begre & Konig, 2008). As SCZ is an exclusively human disorder, animal models cannot mimic all SCZ pathophysiology (Stachowiak et al., 2013; Jones et al., 2011). Some curative animal model studies have failed in human clinical trials (Thomsen et al., 2010; Franco & Cedazo-Minguez, 2014). Thus, it is crucial to develop a novel human-based specific model of SCZ to elucidate mechanisms of the occurrence of the disease to find a neoteric therapy. First time in 2007, Yamanaka et al. reprogrammed human fibroblasts into induced pluripotent stem (iPS) (Takahashi et al., 2007). These cells could be cultured on mouse embryonic fibroblast as feeders (Ellerstrom et al., 2006; Takahashi et al., 2007; Crook et al., 2007). Sun et al. used Matrigel as a feeder-free culture condition to generate individual-specific hiPSCs from the autologous source of cells (Sun et al., 2009). Both human and mouse iPS cells could transform into three germ layers (Maherali et al., 2007), (Meissner et al., 2007; Takahashi et al., 2007; Wernig et al., 2007). HiPSCs develop a source of cells genetically similar to the original cell and are useful for tissue regeneration and drug screening. However, applying viruses to produce hiPSCs has shown a high rate of conveyance utility (Takahashi et al., 2007; Yu et al., 2007). Viruses can also induce a viral reservoir into the host chromosome. Scientists use non-integrating episomal vectors to produce hiPSCs to keep away from accidental genomic pools (Yu et al., 2009).
Considering the importance of developing new approaches to treat SCZ, we aimed to produce hiPSCs in SCZ patients using episomal vectors to evaluate the treatment efficacy of this method in our future studies.
2. Materials and Methods
Preparation of human dermal fibroblasts (HDFs)
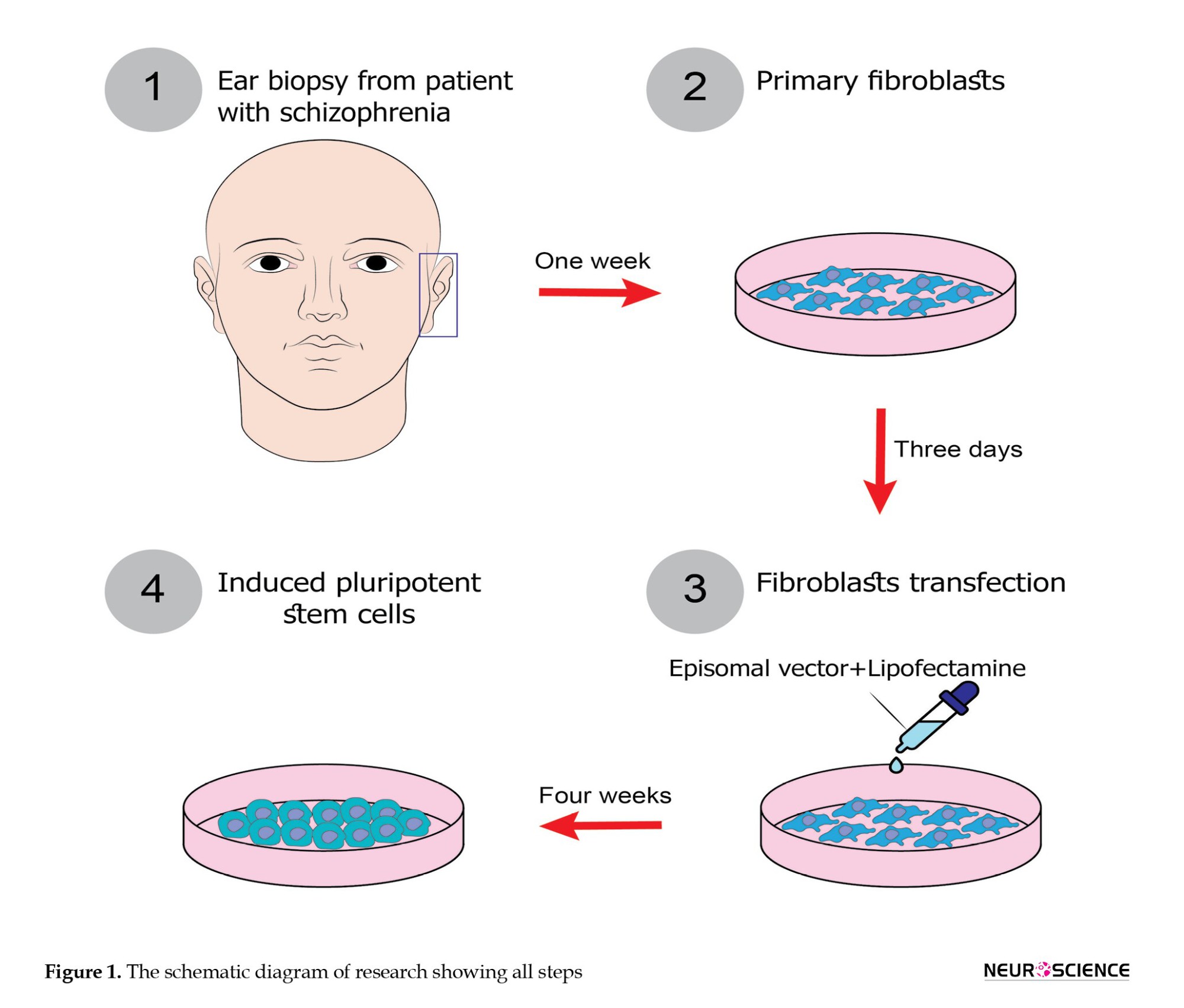
Human skin biopsies were obtained from SCZ patients through dermal punch biopsies after receiving approval from the ethical committee of Iran University of Medical Sciences, Tehran, Iran. The diagnosis of SCZ was confirmed by psychiatrists’ assessment according to the SCID-I criteria. All participants were patients at the Psychiatry Center of Iran. The earlobe tissues (4 mm) were collected, preserved in phosphate-buffered saline (PBS; Medicago, Canada) with 2% pen/strep (Gibco, USA), and transported to the cell culture laboratory at Iran University of Medical Sciences. Tissues were minced and then centrifuged for five minutes (1200 rpm). The supernatant was discarded, and the tissues were digested overnight with Dispase ΙΙ (sigma; USA). Subsequently, enzyme activity was neutralized with DMEM (Gibco, USA) containing 10% FBS (Gibco, USA), and the mixture was centrifuged for five minutes at 1200 rpm. After discarding the supernatant, the tissues were incubated for 30 minutes with collagenase I (Sigma, USA). The collagenase activity was then neutralized as described above. The suspension was filtered through a 70 µm filter (SPL, China) and centrifuged for five minutes at 1200 rpm. The supernatant was removed, and the cell pellets were cultured in 6-well plates containing DMEM with 10% FBS and 1% pen/strep antibiotics (PAN Biotech, Germany), and incubated at 37°C in an atmosphere of 5% CO2. Three dermal biopsies were used in this study. Generation of dermal fibroblast-derived hiPSCs from SCZ patients.
In this study, hiPSCs were generated from dermal fibroblasts using episomal vectors (pEP4 E02S CK2M EN2L, Addgene) containing six factors (OCT-4, SOX-2, KLF-4, c-MYC, Nanog, LIN-28). Approximately 5000 human dermal fibroblasts (HDFs) per cm2 were seeded in each well of a 12-well plate and transfected once using Lipofectamine (Invitrogen, L3000-001). On day 0 or 1, when fibroblasts reached 70-90% confluency, they were transfected according to the kit instructions. Initially, 2.5 µg/µL of episomal vectors and 5 µL of Lipofectamine were mixed for 30 minutes in a microtube, then the mixture was added to the cell culture. After 12 hours, the media was replaced.
Six days later, colonies were transferred onto plates coated with Matrigel (1:30; Sigma, USA) containing DMEM/F12, 20% Knockout Serum Replacement, 100 µM non-essential amino acids, 1% penicillin/streptomycin, 2 mM L-glutamine (all from Gibco, USA), 100 µM ß-mercaptoethanol (Sigma, USA), and 10 ng/ml human basic fibroblast growth factor (b-FGF; PeproTech, USA). The plates were incubated at 5% CO2 with 95% of humidity. To monitor the transfection efficiency, only 6 columns were used for hiPSCs reprogramming, with the remaining serving as controls. After 4 to 5 weeks, colonies were visible. For passaging, colonies were rinsed with PBS and then incubated with DMEM/F12 containing collagenase I (Sigma, USA) at 37°C for 30 minutes. Then, the enzyme was removed, and the plates were rinsed with PBS. Colonies were gently dislodged mechanically and then transferred to new 24-well plates coated with Matrigel. These samples underwent immunocytochemical analysis to assess the expression of Tra1-60, Nanog, Oct3/4, SSEA4, and alkaline phosphatase staining, with at least three clones tested per patient.The schematic diagram of research is shown in Figure 1.
Characterization of the established hiPSCs
Immunocytochemistry (ICC)
To assess the expression of pluripotency markers, colonies were fixed for twenty minutes using 4% paraformaldehyde, then permeabilized with 0.1–0.2% Triton X-100 for 30 minutes. Blocking was performed using 10% mouse serum in PBS for 1 hour at 37°C. For SSEA-4 and TRA-1-60 expression analysis, cells were incubated with Anti-SSEA-4 PE (1:250; Cat. No. CS204438) and Anti-TRA-1-60 FITC (1:200; Cat. No. CS204460) antibodies for 30 minutes at 37°C in a 5% CO2 chamber.
For Oct-3/4 and Nanog expression analysis, the Human Pluripotent Stem Cell 3-Color Immunocytochemistry Kit (Cat No. SC021; bio-techne, USA) was used. Briefly, blocking was done with 10% normal donkey serum in 0.3% Triton® X-100. After removing the blocking buffer, cells were stained with NL637-conjugated Goat Anti-Human Oct-3/4 and NL493-conjugated Goat Anti-Human Nanog antibodies.
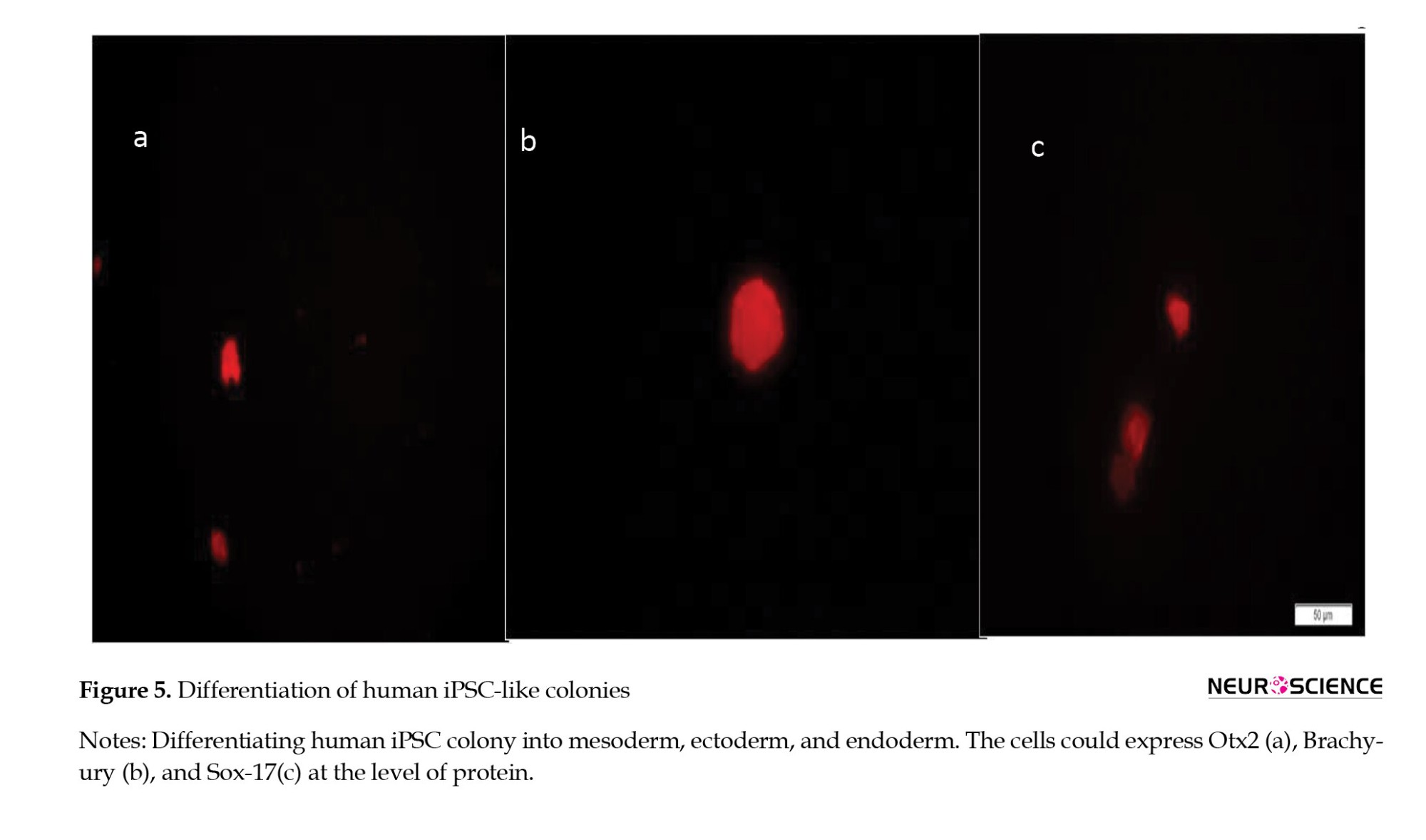
Derived HiPSCs were differentiated into three germ layers. Differentiation was verified using the Human Pluripotent Stem Cell Functional Identification Kit (Cat. No. SC027B), following the manufacturer’s instructions. We used goat anti-human Otx2, goat anti-human Brachyury, and goat anti-human Sox17 as primary antibodies to confirm differentiation into ectoderm, mesoderm, and endoderm, respectively. NL557-conjugated donkey anti-goat (R&D systems, Cat.No.NL001) served as the secondary antibody (red). All nuclei were counterstained with DAPI (blue).
Alkaline phosphatase test
The alkaline phosphatase test was conducted using the Alkaline Phosphatase Staining Kit II (Reprocell, USA) to evaluate AP expression in hiPSC colonies. Briefly, cells were washed with PBST (PBS+triton, final concentration: 0.05%). Then, they were fixed in fixative solution for 2-5 minutes and then incubated in freshly prepared AP solution for 15 minutes at room temperature. The reaction was halted when a bright color developed.
Karyotype analysis
Cells underwent karyotype analysis by being cultured with thymidine (Sigma, USA) for 16 hours at 37°C in 5% CO2. Three hours after removal, cells were exposed to colcemid (Gibco, 0.15 μg/ml, 30 minutes), followed by treatment with 0.075 M KCl at 37°C for 16 minutes. Cells were then fixed three times in ice-cold 3:1 methanol: Glacial acetic acid and dropped onto pre-cleaned, chilled slides. At least 20 metaphase spreads were screened, and 10 were evaluated for chromosomal rearrangements.
Differentiation of hiPSCs into three germ layers
To confirm the pluripotency of hiPSC clones, three lineage differentiation assays were performed using the Human Pluripotent Stem Cell Functional Identification Kit (Cat. No. SC027B). Cells were harvested and prepared for analysis of lineage-specific markers on day 5 (for mesoderm and endoderm lineages) and day 7 (for the ectoderm lineage), as per the manufacturer’s instructions. Immunofluorescence assay was carried out with antibodies to lineage-specific markers for endoderm, ectoderm, and mesoderm. Goat anti-human Otx2, goat anti-human Brachyury, and goat anti-human Sox17 were used as antibodies to verify ectoderm, mesoderm, and endoderm differentiation. Also, we used NL557-conjugated donkey anti-goat (R&D systems, Cat.No.NL001) as the secondary antibody (red). All nuclei were counterstained with DAPI (blue). RNA isolation and quantitative RT-PCR
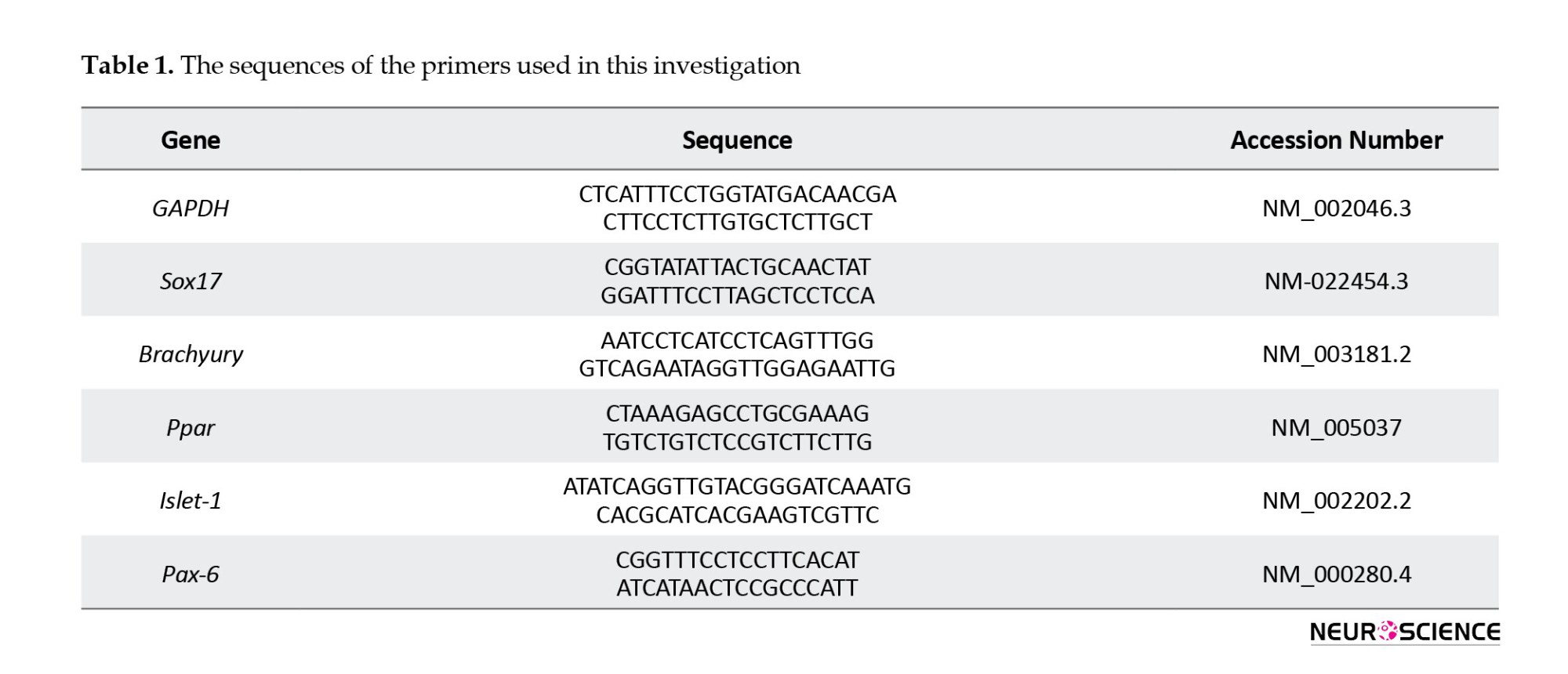
Total RNA was isolated using TRIzol and treated with DNase I to remove genomic DNA contamination. Per the manufacturer’s instructions, 2 µg of total RNA was used for the reverse transcription reaction with the RevertAid First Strand cDNA Synthesis Kit (Fermentas) using an oligo (dt) primer. Quantitative PCR reactions were conducted using Power SYBR Green Master Mix (Applied Biosystems), and results were analyzed on a 7500 real-time PCR system (Applied Biosystems). Gene expression levels were normalized to GAPDH, serving as an internal control, and compared with the same target gene in human dermal fibroblasts (HDF). The primer sequences are listed in Table 1.
Statistical analysis
Data were analyzed by non-parametric Mann-Whitney test using SPSS software, version 16 and P<0.05 were considered significant. Data were presented as Mean±SD.
3. Results
Generation of hiPSCs from dermal fibroblasts of patients with schizophrenia
Isolation and proliferation of human dermal fibroblasts
Human skin biopsies were cultured in 6-well plates (Figure 2a). Spindle-shaped cells migrated from the biopsies within 4-5 weeks of culture. Then, biopsies were removed, and cells were allowed to reach approximately 75% confluence over 14 days, with medium changes every other day (Figure 2b).

Characterization of established hiPSCs
Immunocytochemistry, ALP test, and karyotype analysis
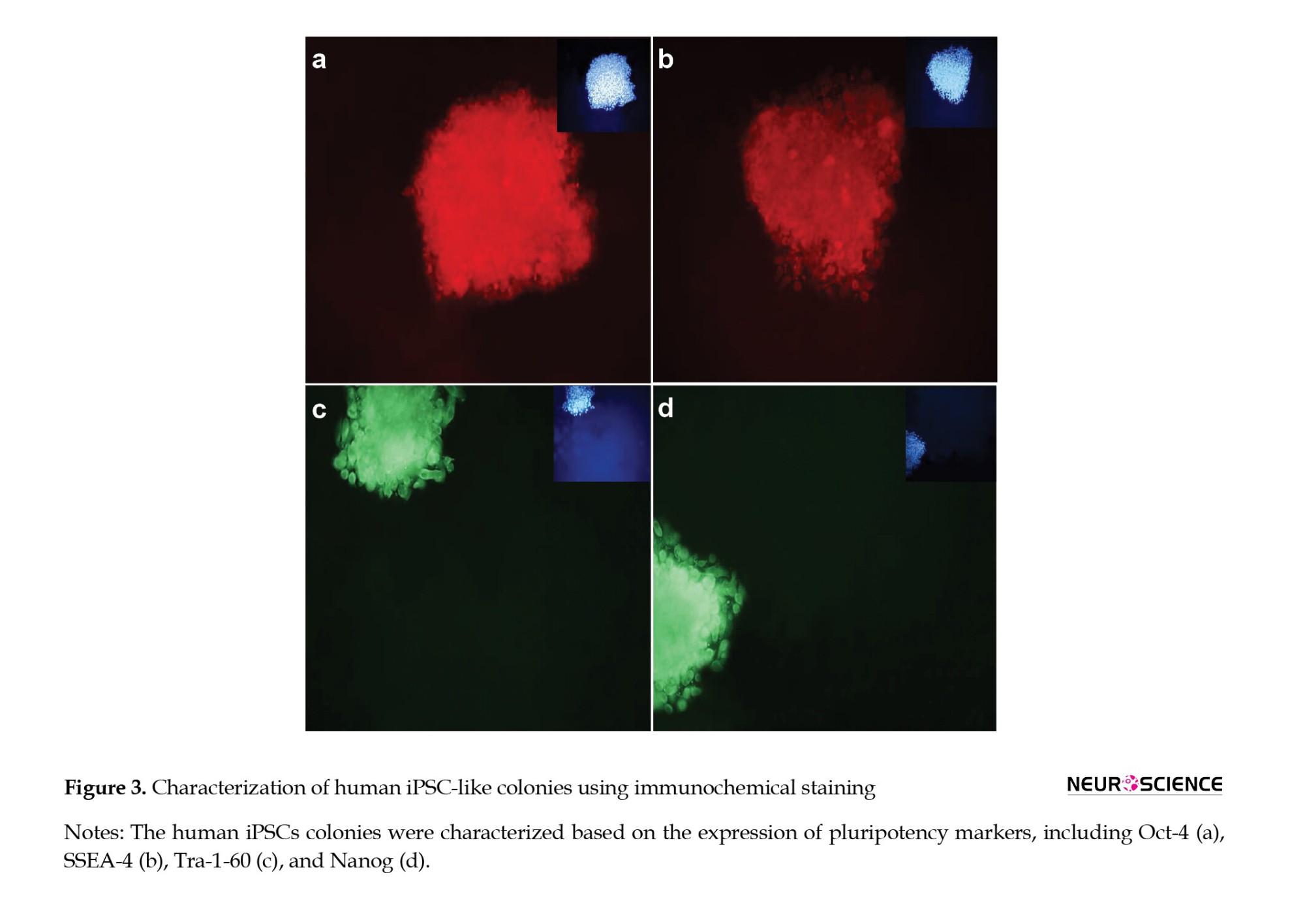
After introducing reprogramming factors, cells began to form colonies with hESC-like morphology (Figure 2c), which exhibited intense ALP activity (Figure 2d). These colonies were analyzed via immunocytochemistry and gene expression assays, expressing pluripotent markers including Tra-1-60, Oct4, SSEA4, and Nanog at the protein level (Figure 3).
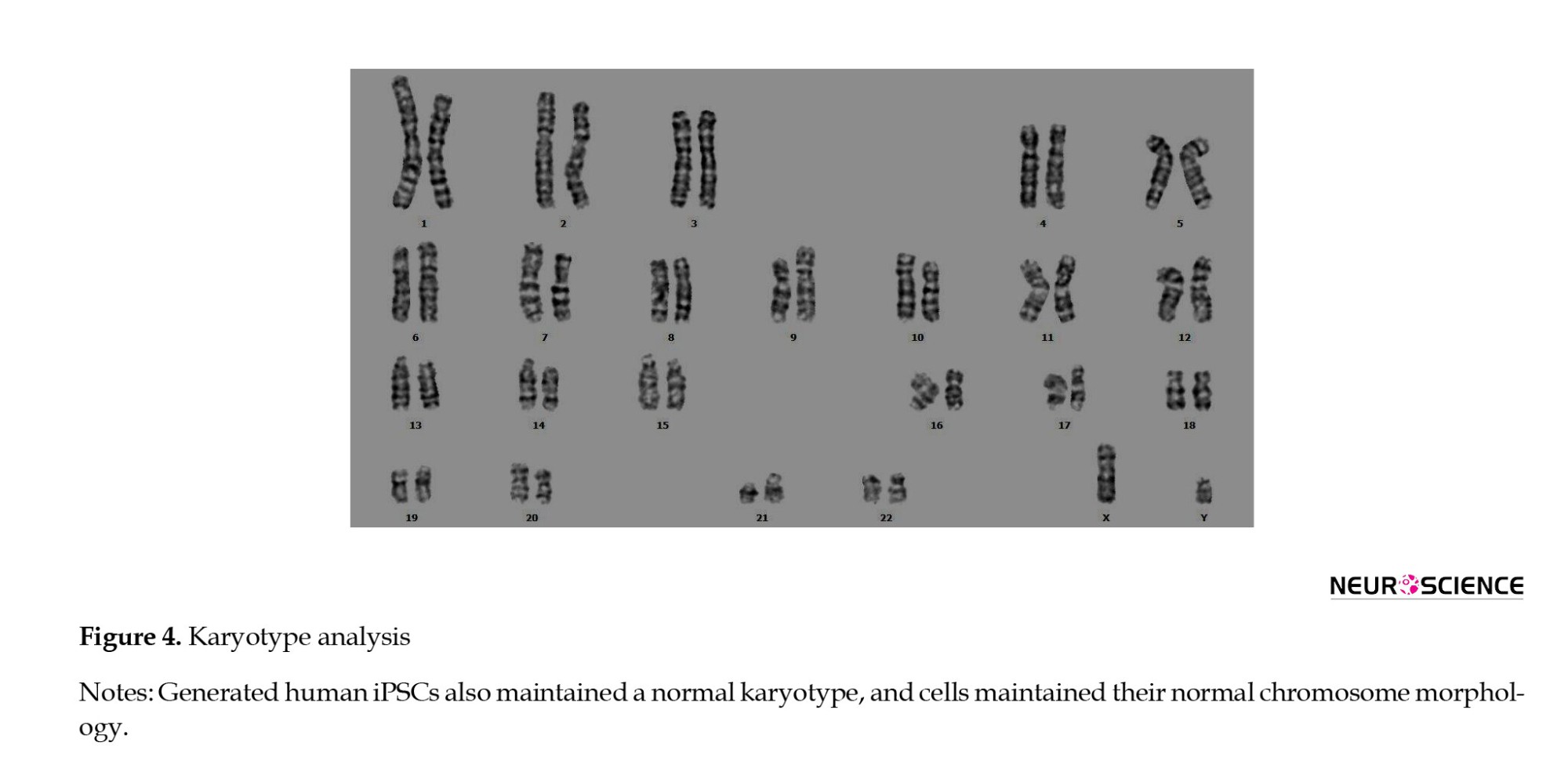
The putative hiPSCs maintained a normal karyotype (Figure 4).
Differentiation into three germ layers
The generated cells were continuously cultured with weekly passaging at a split ratio of 1:3. To confirm their multilineage differentiation potential, cells were induced to differentiate into ectoderm, mesoderm, and endoderm. Immunocytochemical staining demonstrated expression of Otx2, Brachyury, and Sox-17, markers of ectoderm, mesoderm, and endoderm, respectively (Figure 5).
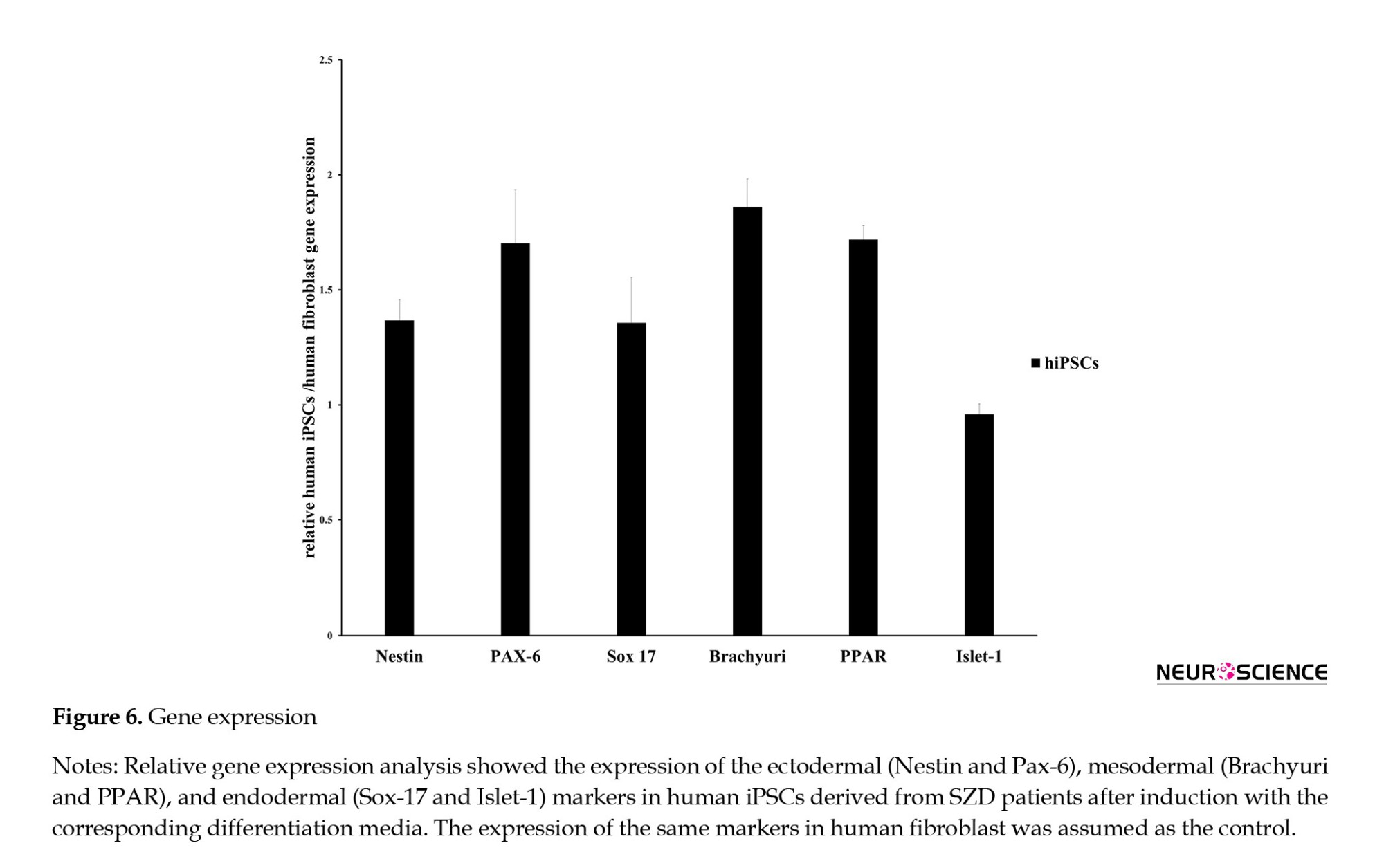
Gene expression was assessed for ectoderm differentiation markers Nestin and Pax-6; mesoderm markers Brachyury and PPAR; and endoderm markers Islet-1 and Sox-17. The differentiated hiPSCs showed upregulated gene expression compared to fibroblasts (Figure 6).
4. Discussion
SCZ is defined as a neurodevelopmental disorder that alters the processes of thought, perception, and emotion, leading to mental deterioration. In the current study, we generated human induced pluripotent stem cells (hiPSCs) from dermal fibroblasts of patients with SCZD using episomal vectors on Matrigel-coated plates. Our results demonstrated that the derived colonies could express pluripotent markers at the protein level, including SSEA-4, Oct-4, Nanog, and Tra-1-60. The generated hiPSCs maintained their normal karyotype, and we could also detect strong ALP expression in the colonies. After induction, the cells could express ectodermal (Nestin), endodermal (Islet-1, Sox-17), and mesodermal (Brachyury and PPAR) markers at the level of mRNA when the results were compared with fibroblasts confirming the pluripotent properties of the generated cells. Previous studies have shown that iPSCs can be cultured on mouse embryonic fibroblast feeders (Ellerstrom et al., 2006; Takahashi et al., 2007; Crook et al., 2007). Alternatively, feeders feeders can be replaced by Matrigel, as we have done in our project (Kleinman & Martin 2005). In an investigation, Sun et al. used Matrigel as a feeder-free culture condition to generate individual-specific hiPSCs from an autologous source of cells (Sun et al., 2009). Moreover, using conditioned media and Matrigel supported with essential growth factors could reduce the contamination of stem cells (Ghasemi-Dehkordi et al., 2015). The breaking points for hiPSCs for investigating brain disturbance are neuron and patient variability. This variation may be due to differences in the type of viruses used, epigenetic factors, spontaneous mutation, differences in techniques, and cell type of origin. Some scientists have investigated the advantages and difficulties of applying viral integration methods versus episomal or other methods. Viral methods induce genomic deviation (Hussein et al., 2011). They are more efficient than other methods consisting of recombinant protein (Zhou et al., 2009), miRNA (Miyoshi et al., 2011), and mRNA (Warren et al., 2010). Most SCZ patients show different responses to different treatments (Schennach et al., 2012), and only a few percent effectively respond to the therapy (Emsley et al., 2011). The application of new methods for treating this disease is critical.
Conclusion
The use of hiPSCs derived from patients with schizophrenia could be a promising approach for treating the disease and screening drugs. Further studies are suggested to investigate the behavior of these cells during neurogenesis.
Ethical Considerations
Compliance with ethical guidelines
There were no ethical considerations to be considered in this research.
Funding
This project was pecuniary reinforcement by the Iran University of Medical Sciences (Code: 96-02-87-31209).
Authors' contributions
All authors equally contributed to preparing this article.
Conflict of interest
The authors declared no conflict of interest.
References
Schizophrenia (SCZ) is a severe neuropsychiatric disorder with a global prevalence of 1% and 80-85% heritability (Sullivan et al., 2003). Multiple reasons, including genetic and environmental factors, have roles in the pathogenesis of SCZ (Tsuang et al., 2001; Palha & Goodman, 2006), and perturbation of these agents could cause the disturbance of information of the neural network and brain function (Christian et al., 2010). SCZ onset often happens in early adulthood (Sawa & Snyder, 2002), but in some patients, the abnormal neurodevelopmental process may start in childhood (Weinberger, 1987; Marin, 2012). Furthermore, most patients show different responses to treatment (Schennach et al., 2012), and only a few percent effectively respond to the treatment (Emsley et al., 2011). Studies have shown that SCZ is accompanied by abnormal neuronal communication with a significant role for oligodendrocyte and progenitor cells in the pathogenesis (Rubinov, 2013; Federspiel et al., 2006; Begre & Konig, 2008). As SCZ is an exclusively human disorder, animal models cannot mimic all SCZ pathophysiology (Stachowiak et al., 2013; Jones et al., 2011). Some curative animal model studies have failed in human clinical trials (Thomsen et al., 2010; Franco & Cedazo-Minguez, 2014). Thus, it is crucial to develop a novel human-based specific model of SCZ to elucidate mechanisms of the occurrence of the disease to find a neoteric therapy. First time in 2007, Yamanaka et al. reprogrammed human fibroblasts into induced pluripotent stem (iPS) (Takahashi et al., 2007). These cells could be cultured on mouse embryonic fibroblast as feeders (Ellerstrom et al., 2006; Takahashi et al., 2007; Crook et al., 2007). Sun et al. used Matrigel as a feeder-free culture condition to generate individual-specific hiPSCs from the autologous source of cells (Sun et al., 2009). Both human and mouse iPS cells could transform into three germ layers (Maherali et al., 2007), (Meissner et al., 2007; Takahashi et al., 2007; Wernig et al., 2007). HiPSCs develop a source of cells genetically similar to the original cell and are useful for tissue regeneration and drug screening. However, applying viruses to produce hiPSCs has shown a high rate of conveyance utility (Takahashi et al., 2007; Yu et al., 2007). Viruses can also induce a viral reservoir into the host chromosome. Scientists use non-integrating episomal vectors to produce hiPSCs to keep away from accidental genomic pools (Yu et al., 2009).
Considering the importance of developing new approaches to treat SCZ, we aimed to produce hiPSCs in SCZ patients using episomal vectors to evaluate the treatment efficacy of this method in our future studies.
2. Materials and Methods
Preparation of human dermal fibroblasts (HDFs)
Human skin biopsies were obtained from SCZ patients through dermal punch biopsies after receiving approval from the ethical committee of Iran University of Medical Sciences, Tehran, Iran. The diagnosis of SCZ was confirmed by psychiatrists’ assessment according to the SCID-I criteria. All participants were patients at the Psychiatry Center of Iran. The earlobe tissues (4 mm) were collected, preserved in phosphate-buffered saline (PBS; Medicago, Canada) with 2% pen/strep (Gibco, USA), and transported to the cell culture laboratory at Iran University of Medical Sciences. Tissues were minced and then centrifuged for five minutes (1200 rpm). The supernatant was discarded, and the tissues were digested overnight with Dispase ΙΙ (sigma; USA). Subsequently, enzyme activity was neutralized with DMEM (Gibco, USA) containing 10% FBS (Gibco, USA), and the mixture was centrifuged for five minutes at 1200 rpm. After discarding the supernatant, the tissues were incubated for 30 minutes with collagenase I (Sigma, USA). The collagenase activity was then neutralized as described above. The suspension was filtered through a 70 µm filter (SPL, China) and centrifuged for five minutes at 1200 rpm. The supernatant was removed, and the cell pellets were cultured in 6-well plates containing DMEM with 10% FBS and 1% pen/strep antibiotics (PAN Biotech, Germany), and incubated at 37°C in an atmosphere of 5% CO2. Three dermal biopsies were used in this study. Generation of dermal fibroblast-derived hiPSCs from SCZ patients.
In this study, hiPSCs were generated from dermal fibroblasts using episomal vectors (pEP4 E02S CK2M EN2L, Addgene) containing six factors (OCT-4, SOX-2, KLF-4, c-MYC, Nanog, LIN-28). Approximately 5000 human dermal fibroblasts (HDFs) per cm2 were seeded in each well of a 12-well plate and transfected once using Lipofectamine (Invitrogen, L3000-001). On day 0 or 1, when fibroblasts reached 70-90% confluency, they were transfected according to the kit instructions. Initially, 2.5 µg/µL of episomal vectors and 5 µL of Lipofectamine were mixed for 30 minutes in a microtube, then the mixture was added to the cell culture. After 12 hours, the media was replaced.
Six days later, colonies were transferred onto plates coated with Matrigel (1:30; Sigma, USA) containing DMEM/F12, 20% Knockout Serum Replacement, 100 µM non-essential amino acids, 1% penicillin/streptomycin, 2 mM L-glutamine (all from Gibco, USA), 100 µM ß-mercaptoethanol (Sigma, USA), and 10 ng/ml human basic fibroblast growth factor (b-FGF; PeproTech, USA). The plates were incubated at 5% CO2 with 95% of humidity. To monitor the transfection efficiency, only 6 columns were used for hiPSCs reprogramming, with the remaining serving as controls. After 4 to 5 weeks, colonies were visible. For passaging, colonies were rinsed with PBS and then incubated with DMEM/F12 containing collagenase I (Sigma, USA) at 37°C for 30 minutes. Then, the enzyme was removed, and the plates were rinsed with PBS. Colonies were gently dislodged mechanically and then transferred to new 24-well plates coated with Matrigel. These samples underwent immunocytochemical analysis to assess the expression of Tra1-60, Nanog, Oct3/4, SSEA4, and alkaline phosphatase staining, with at least three clones tested per patient.The schematic diagram of research is shown in Figure 1.
Characterization of the established hiPSCs
Immunocytochemistry (ICC)
To assess the expression of pluripotency markers, colonies were fixed for twenty minutes using 4% paraformaldehyde, then permeabilized with 0.1–0.2% Triton X-100 for 30 minutes. Blocking was performed using 10% mouse serum in PBS for 1 hour at 37°C. For SSEA-4 and TRA-1-60 expression analysis, cells were incubated with Anti-SSEA-4 PE (1:250; Cat. No. CS204438) and Anti-TRA-1-60 FITC (1:200; Cat. No. CS204460) antibodies for 30 minutes at 37°C in a 5% CO2 chamber.
For Oct-3/4 and Nanog expression analysis, the Human Pluripotent Stem Cell 3-Color Immunocytochemistry Kit (Cat No. SC021; bio-techne, USA) was used. Briefly, blocking was done with 10% normal donkey serum in 0.3% Triton® X-100. After removing the blocking buffer, cells were stained with NL637-conjugated Goat Anti-Human Oct-3/4 and NL493-conjugated Goat Anti-Human Nanog antibodies.
Derived HiPSCs were differentiated into three germ layers. Differentiation was verified using the Human Pluripotent Stem Cell Functional Identification Kit (Cat. No. SC027B), following the manufacturer’s instructions. We used goat anti-human Otx2, goat anti-human Brachyury, and goat anti-human Sox17 as primary antibodies to confirm differentiation into ectoderm, mesoderm, and endoderm, respectively. NL557-conjugated donkey anti-goat (R&D systems, Cat.No.NL001) served as the secondary antibody (red). All nuclei were counterstained with DAPI (blue).
Alkaline phosphatase test
The alkaline phosphatase test was conducted using the Alkaline Phosphatase Staining Kit II (Reprocell, USA) to evaluate AP expression in hiPSC colonies. Briefly, cells were washed with PBST (PBS+triton, final concentration: 0.05%). Then, they were fixed in fixative solution for 2-5 minutes and then incubated in freshly prepared AP solution for 15 minutes at room temperature. The reaction was halted when a bright color developed.
Karyotype analysis
Cells underwent karyotype analysis by being cultured with thymidine (Sigma, USA) for 16 hours at 37°C in 5% CO2. Three hours after removal, cells were exposed to colcemid (Gibco, 0.15 μg/ml, 30 minutes), followed by treatment with 0.075 M KCl at 37°C for 16 minutes. Cells were then fixed three times in ice-cold 3:1 methanol: Glacial acetic acid and dropped onto pre-cleaned, chilled slides. At least 20 metaphase spreads were screened, and 10 were evaluated for chromosomal rearrangements.
Differentiation of hiPSCs into three germ layers
To confirm the pluripotency of hiPSC clones, three lineage differentiation assays were performed using the Human Pluripotent Stem Cell Functional Identification Kit (Cat. No. SC027B). Cells were harvested and prepared for analysis of lineage-specific markers on day 5 (for mesoderm and endoderm lineages) and day 7 (for the ectoderm lineage), as per the manufacturer’s instructions. Immunofluorescence assay was carried out with antibodies to lineage-specific markers for endoderm, ectoderm, and mesoderm. Goat anti-human Otx2, goat anti-human Brachyury, and goat anti-human Sox17 were used as antibodies to verify ectoderm, mesoderm, and endoderm differentiation. Also, we used NL557-conjugated donkey anti-goat (R&D systems, Cat.No.NL001) as the secondary antibody (red). All nuclei were counterstained with DAPI (blue). RNA isolation and quantitative RT-PCR
Total RNA was isolated using TRIzol and treated with DNase I to remove genomic DNA contamination. Per the manufacturer’s instructions, 2 µg of total RNA was used for the reverse transcription reaction with the RevertAid First Strand cDNA Synthesis Kit (Fermentas) using an oligo (dt) primer. Quantitative PCR reactions were conducted using Power SYBR Green Master Mix (Applied Biosystems), and results were analyzed on a 7500 real-time PCR system (Applied Biosystems). Gene expression levels were normalized to GAPDH, serving as an internal control, and compared with the same target gene in human dermal fibroblasts (HDF). The primer sequences are listed in Table 1.
Statistical analysis
Data were analyzed by non-parametric Mann-Whitney test using SPSS software, version 16 and P<0.05 were considered significant. Data were presented as Mean±SD.
3. Results
Generation of hiPSCs from dermal fibroblasts of patients with schizophrenia
Isolation and proliferation of human dermal fibroblasts
Human skin biopsies were cultured in 6-well plates (Figure 2a). Spindle-shaped cells migrated from the biopsies within 4-5 weeks of culture. Then, biopsies were removed, and cells were allowed to reach approximately 75% confluence over 14 days, with medium changes every other day (Figure 2b).
Characterization of established hiPSCs
Immunocytochemistry, ALP test, and karyotype analysis
After introducing reprogramming factors, cells began to form colonies with hESC-like morphology (Figure 2c), which exhibited intense ALP activity (Figure 2d). These colonies were analyzed via immunocytochemistry and gene expression assays, expressing pluripotent markers including Tra-1-60, Oct4, SSEA4, and Nanog at the protein level (Figure 3).
The putative hiPSCs maintained a normal karyotype (Figure 4).
Differentiation into three germ layers
The generated cells were continuously cultured with weekly passaging at a split ratio of 1:3. To confirm their multilineage differentiation potential, cells were induced to differentiate into ectoderm, mesoderm, and endoderm. Immunocytochemical staining demonstrated expression of Otx2, Brachyury, and Sox-17, markers of ectoderm, mesoderm, and endoderm, respectively (Figure 5).
Gene expression was assessed for ectoderm differentiation markers Nestin and Pax-6; mesoderm markers Brachyury and PPAR; and endoderm markers Islet-1 and Sox-17. The differentiated hiPSCs showed upregulated gene expression compared to fibroblasts (Figure 6).
4. Discussion
SCZ is defined as a neurodevelopmental disorder that alters the processes of thought, perception, and emotion, leading to mental deterioration. In the current study, we generated human induced pluripotent stem cells (hiPSCs) from dermal fibroblasts of patients with SCZD using episomal vectors on Matrigel-coated plates. Our results demonstrated that the derived colonies could express pluripotent markers at the protein level, including SSEA-4, Oct-4, Nanog, and Tra-1-60. The generated hiPSCs maintained their normal karyotype, and we could also detect strong ALP expression in the colonies. After induction, the cells could express ectodermal (Nestin), endodermal (Islet-1, Sox-17), and mesodermal (Brachyury and PPAR) markers at the level of mRNA when the results were compared with fibroblasts confirming the pluripotent properties of the generated cells. Previous studies have shown that iPSCs can be cultured on mouse embryonic fibroblast feeders (Ellerstrom et al., 2006; Takahashi et al., 2007; Crook et al., 2007). Alternatively, feeders feeders can be replaced by Matrigel, as we have done in our project (Kleinman & Martin 2005). In an investigation, Sun et al. used Matrigel as a feeder-free culture condition to generate individual-specific hiPSCs from an autologous source of cells (Sun et al., 2009). Moreover, using conditioned media and Matrigel supported with essential growth factors could reduce the contamination of stem cells (Ghasemi-Dehkordi et al., 2015). The breaking points for hiPSCs for investigating brain disturbance are neuron and patient variability. This variation may be due to differences in the type of viruses used, epigenetic factors, spontaneous mutation, differences in techniques, and cell type of origin. Some scientists have investigated the advantages and difficulties of applying viral integration methods versus episomal or other methods. Viral methods induce genomic deviation (Hussein et al., 2011). They are more efficient than other methods consisting of recombinant protein (Zhou et al., 2009), miRNA (Miyoshi et al., 2011), and mRNA (Warren et al., 2010). Most SCZ patients show different responses to different treatments (Schennach et al., 2012), and only a few percent effectively respond to the therapy (Emsley et al., 2011). The application of new methods for treating this disease is critical.
Conclusion
The use of hiPSCs derived from patients with schizophrenia could be a promising approach for treating the disease and screening drugs. Further studies are suggested to investigate the behavior of these cells during neurogenesis.
Ethical Considerations
Compliance with ethical guidelines
There were no ethical considerations to be considered in this research.
Funding
This project was pecuniary reinforcement by the Iran University of Medical Sciences (Code: 96-02-87-31209).
Authors' contributions
All authors equally contributed to preparing this article.
Conflict of interest
The authors declared no conflict of interest.
References
Begré, S., & Koenig, T. (2008). Cerebral disconnectivity: an early event in schizophrenia. The Neuroscientist: A review Journal Bringing Neurobiology, Neurology and Psychiatry, 14(1), 19–45.[DOI:10.1177/1073858406298391] [PMID]
Christian, K., Song, H., & Ming, G. L. (2010). Adult neurogenesis as a cellular model to study schizophrenia. Cell Cycle (Georgetown, Tex.), 9(4), 636–637. [DOI:10.4161/cc.9.4.10932] [PMID]
Crook, J. M., Peura, T. T., Kravets, L., Bosman, A. G., Buzzard, J. J., & Horne, R., et al. (2007). The generation of six clinical-grade human embryonic stem cell lines. Cell Stem Cell, 1(5), 490–494. [DOI:10.1016/j.stem.2007.10.004] [PMID]
Ellerstrom, C., Strehl, R., Moya, K., Andersson, K., Bergh, C., & Lundin, K., et al. (2006). “Derivation of a xeno-free human embryonic stem cell line.” Stem Cells (Dayton, Ohio), 24(10), 2170–2176. [DOI:10.1634/stemcells.2006-0130] [PMID]
Emsley, R., Chiliza, B., Asmal, L., & Lehloenya, K. (2011). The concepts of remission and recovery in schizophrenia. Current Opinion in Psychiatry, 24(2), 114–121. [DOI:10.1097/YCO.0b013e3283436ea3] [PMID]
Federspiel, A., Begré, S., Kiefer, C., Schroth, G., Strik, W. K., & Dierks, T. (2006). Alterations of white matter connectivity in first-episode schizophrenia.” Neurobiology of Disease, 22(3), 702-709. [DOI:10.1016/j.nbd.2006.01.015] [PMID]
Franco, R., & Cedazo-Minguez, A. (2014). Successful therapies for Alzheimer’s disease: Why so many in animal models and none in humans? Frontiers in Pharmacology, 5, 146. [DOI:10.3389/fphar.2014.00146]
Ghasemi-Dehkordi, P., Allahbakhshian-Farsani, M., Abdian, N., Mirzaeian, A., Hashemzadeh-Chaleshtori, M., & Jafari-Ghahfarokhi, H. (2015). “Effects of feeder layers, culture media, conditional media, growth factors, and passage number on stem cell optimization.” Proceedings of the National Academy of Sciences, India Section B: Biological Sciences, 85(3), 711-717. [DOI:10.1007/s40011-014-0430-8]
Hussein, S. M., Nagy, K., & Nagy, A. (2011). Human- induced pluripotent stem cells: The past, present, and future. Clinical Pharmacology & Therapeutics, 89(5), 741-745. [DOI:10.1038/clpt.2011.37] [PMID]
Jones, C. A., Watson, D. J., & Fone, K. C. (2011). Animal models of schizophrenia.” British Journal of Pharmacology, 164(4), 1162-1194. [DOI:10.1111/j.1476-5381.2011.01386.x] [PMID] [PMCID]
Kleinman, H. K., & Martin, G. R. (2005). Matrigel: Basement membrane matrix with biological activity. Seminars in Cancer Biology, 15(5), 378–386. [DOI:10.1016/j.semcancer.2005.05.004] [PMID]
Maherali, N., Sridharan, R., Xie, W., Utikal, J., Eminli, S., & Arnold, K., et al. (2007). “Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution.” Cell Stem Cell, 1(1), 55–70. [DOI:10.1016/j.stem.2007.05.014] [PMID]
Marin, O. (2012). “Interneuron dysfunction in psychiatric disorders.” Nature Reviews. Neuroscience, 13(2), 107–120.[DOI:10.1038/nrn3155] [PMID]
Meissner, A., Wernig, M., & Jaenisch, R. (2007). Direct reprogramming of genetically unmodified fibroblasts into pluripotent stem cells. Nature Biotechnology, 25(10), 1177–1181.[DOI:10.1038/nbt1335] [PMID]
Miyoshi, N., Ishii, H., Nagano, H., Haraguchi, N., Dewi, D. L., & Kano, Y., et al. (2011). “Reprogramming of mouse and human cells to pluripotency using mature microRNAs.”Cell Stem Cell, 8(6), 633–638. [DOI:10.1016/j.stem.2011.05.001] [PMID]
Palha, J. A., & Goodman, A. B. (2006). “Thyroid hormones and retinoids: A possible link between genes and environment in schizophrenia.” Brain Research Reviews, 51(1), 61-71. [DOI:10.1016/j.brainresrev.2005.10.001] [PMID]
Rubinov, M., & Bullmore, E. (2013). “Schizophrenia and abnormal brain network hubs.” Dialogues in Clinical Neuroscience, 15(3), 339–349. [PMID]
Sawa, A., & Snyder, S. H. (2002). “Schizophrenia: Diverse approaches to a complex disease.” Science, 296(5568), 692-695. [DOI:10.1126/science.1070532] [PMID]
Schennach, R., Meyer, S., Seemüller, F., Jäger, M., Schmauss, M., & Laux, G., et al. (2012). Response trajectories in "real-world" naturalistically treated schizophrenia patients. Schizophrenia Research, 139(1-3), 218–224. [DOI:10.1016/j.schres.2012.05.004] [PMID]
Stachowiak, M. K., Kucinski, A., Curl, R., Syposs, C., Yang, Y., & Narla, S., et al. (2013). “Schizophrenia: A neurodevelopmental disorder—integrative genomic hypothesis and therapeutic implications from a transgenic mouse model.” Schizophrenia Research, 143(2-3), 367-376. [DOI:10.1016/j.schres.2012.11.004] [PMID]
Sullivan, P. F., Kendler, K. S., & Neale, M. C. (2003). “Schizophrenia is a complex trait: Evidence from a meta-analysis of twin studies.” Archives of General Psychiatry, 60(12), 1187-1192. [DOI:10.1001/archpsyc.60.12.1187] [PMID]
Sun, N., Panetta, N. J., Gupta, D. M., Wilson, K. D., Lee, A., & Jia, F., et al. (2009). “Feeder-free derivation of induced pluripotent stem cells from adult human adipose stem cells.” Proceedings of the National Academy of Sciences of the United States of America, 106(37), 15720–15725. [DOI:10.1073/pnas.0908450106] [PMID] [PMCID]
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., & Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell, 131(5), 861-872. [DOI:10.1016/j.cell.2007.11.019] [PMID]
Thomsen, M. S., Hansen, H. H., Timmerman, D. B., & Mikkelsen, J. D. (2010). Cognitive improvement by activation of α7- nicotinic acetylcholine receptors: From animal models to human pathophysiology.” Current Pharmaceutical Design, 16(3), 323-343. [DOI:10.2174/138161210790170094] [PMID]
Tsuang, M. T., Stone, W. S., & Faraone, S. V. (2001). “Genes, environment and schizophrenia.” The British journal of Psychiatry. Supplement, 40, s18–s24. [DOI:10.1192/bjp.178.40.s18] [PMID]
Warren, L., Manos, P. D., Ahfeldt, T., Loh, Y. H., Li, H., & Lau, F., et al. (2010). “Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA.” Cell Stem Cell, 7(5), 618-630. [DOI:10.1016/j.stem.2010.08.012] [PMID] [PMCID]
Weinberger, D. R. (1987). “Implications of normal brain development for the pathogenesis of schizophrenia.” Archives of General Psychiatry, 44(7), 660-669. [DOI:10.1001/archpsyc.1987.01800190080012] [PMID]
Wernig, M., Meissner, A., Foreman, R., Brambrink, T., Ku, M., & Hochedlinger, K., et al. (2007). “In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state.” Nature 448(7151): 318-324. [DOI:10.1038/nature05944] [PMID]
Type of Study: Original |
Subject:
Cellular and molecular Neuroscience
Received: 2021/09/6 | Accepted: 2022/06/21 | Published: 2024/05/29
Received: 2021/09/6 | Accepted: 2022/06/21 | Published: 2024/05/29
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
