

Volume 15, Issue 1 (January & February 2024)
BCN 2024, 15(1): 1-26 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Thomas J, Wilson S. Molecular and Therapeutic Targets for Amyloid-beta Plaques in Alzheimer's Disease: A Review Study. BCN 2024; 15 (1) :1-26
URL: http://bcn.iums.ac.ir/article-1-2206-en.html
URL: http://bcn.iums.ac.ir/article-1-2206-en.html



1- Department of Pharmacology, School of Pharmacy University of Amrita Vishwavidyapeetham, Guntur, India.
2- University of Amrita Vishwavidyapeetham, Coimbatore, India.
2- University of Amrita Vishwavidyapeetham, Coimbatore, India.
Full-Text [PDF 1823 kb]
| Abstract (HTML)
Full-Text:
1. Introduction
Alzheimer's disease (AD) is a progressive neuronal disorder that develops with age and leads to cognitive decline and thinking skills. AD has been a global concern for years, comprising 60-80% of dementia cases (Ashraf et al., 2016). The Alzheimer's association estimated that 10 million around the globe are affected by AD and the number is on the rise. India is ranked third highest in the caseload related to dementia and particularly in AD after China and the US. According to statistics, the population of the elderly in India will reach around 300 million, accounting for almost 20% of the total population of the country. The expected rise of AD patients in the world by 2050 would be from 1.6 million cases in 2015 to 4.6 million cases by 2050 (Edwards, 2019; Madav et al., 2019; Rizzi et al., 2014; Soto et al., 2007). Pathologically, AD is characterized by intracellular neurofibrillary tangles and extracellular amyloid-beta (Aβ) plaques, which are first seen in the cortex and hippocampus. This induces neuronal injury resulting in neuronal death and successively damaging the cholinergic transmission leading to loss of memory and cognition along with neurotransmitter abnormalities (Benny & Thomas, 2019; Chaudhary et al., 2018; Gopalakrishnan et al., 2019; Shankarappa et al., 2012).
Over the years, advances in the field of pathogenesis have inspired researchers to identify novel therapies concentrated more on the pathological aspects of the disease. Although the exact root cause of AD remains unclear, several factors play a major role in the progression of the disease (Madav et al., 2019). The various hypotheses developed for the pathogenesis of AD are included in Figure 1.

The Aβ hypothesis remains the most studied area of AD pathology and the preferred target for drug development. The proteolytic processing of amyloid precursor protein (APP) is said to be the central dogma of this hypothesis. APP is a transmembrane protein located on chromosome 21 in humans and is expressed in the brain at high levels and is metabolized rapidly. APP is mainly synthesized in the endoplasmic reticulum and then moves to the Golgi network where it plays a role in neural growth and repair. The proteolysis of APP occurs by amyloidogenic (pathogenic) or the non-amyloidogenic (non-pathogenic) pathway. The amyloidogenic pathway releases insoluble Aβ peptides and the aggregation of these peptides leads to plaque formation (Du et al., 2018; Edwards, 2019; Serrano-Pozo et al., 2011; Zhang et al., 2011).
2. Aβ: The miracle worker
Aβ is a peptide generated throughout life, while the formation of Aβ plaques is a neuropathological hallmark of AD stimulated by synaptic activity. Production of the short Aβ peptides is not toxic, but aggregated Aβ fibrils are a pathological condition of AD (Soto et al., 2007). Although early studies have suggested that high Aβ accumulation is the cause of plaque formation and neuronal cell toxicity, they have concluded that even the short insoluble Aβ peptides may be more toxic (Coman & Nemeş, 2017). In a normal brain, an enzyme called α-secretase acts on APP and cleaves into secreted APPα (SAPPα) and an 83 amino acid membrane-bound C-terminal fragment called CTF83. Alternatively, in an AD brain, an enzyme called β-secretase acts on APP and cleaves it into secreted APPβ (SAPPβ) and a 99 amino acid long, membrane-bound C-terminal fragment called CTF99. In a normal brain, CTF83 is further cleaved by the γ-secretase complex and leads to the generation of APP intracellular domain (AICD) fragments, which translocate to the nucleus and regulate the proteins of neuroprotective pathways. However, in AD, γ-secretase cleaves the CTF99 fragment into Aβ40-42 peptides leading to the formation of Aβ fragments. Soluble Aβ fragments are dissolved whereas under certain conditions, insoluble Aβ fragments are aggregated leading to plaque formation and disruption in cell communication (Chaudhary et al., 2018; Du et al., 2018; Edwards, 2019; Octave, 1995) including abnormal deposit of amyloid β (Aβ. In humans, the estimated physiological rate of production of Aβ is 7.6% per hour and clearance rate is 8.3% per hour and proteolytic degradation is the major route of clearance. The targets particularly related to the formation of Aβ plaques have been mentioned below. Few targets directly contribute to Aβ plaques by cleaving APP directly (γ-secretase), whereas the others play their roles indirectly by over expression of apolipoprotein E (APOE), dual-specificity tyrosine-regulated kinases (DYRK1A), nod-like receptor protein 3 (NLRP3), casein kinase 1 (CK1), Cyclin-dependent kinase 5 (Cdk5), triggering receptor expressed on myeloid cells 2 (TREM2) and matrix metallopeptidases (MMP) as mentioned in Table 1.

β-secretase 1 (BACE1)
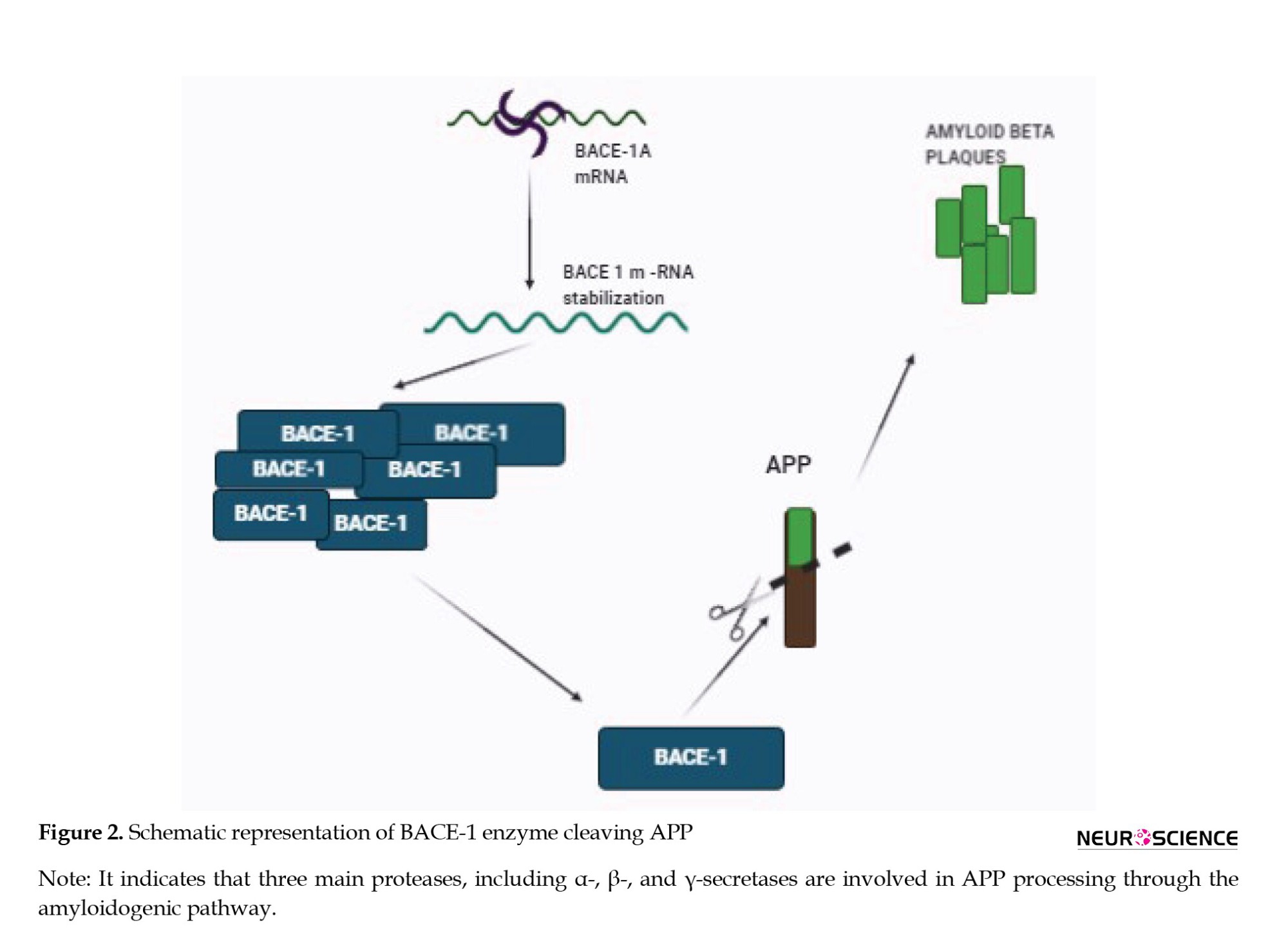
BACE1 was identified in 1999 and belongs to the class of aspartyl proteases with disulfide bond arrangement and consists of 501 amino acids. It is predominantly found in the regions of the brain and is said to be the major determinant of amyloidosis in the CNS (Evin & Hince, 2013). BACE1 is a key enzyme that regulates the β-secretase activity of APP. Mutation in APP leads to the increased access of the enzyme to β-secretase cleavage site and augments Aβ production (Vassar & Kandalepas, 2011). Deposition of Aβ peptides leads to the formation of large Aβ plaques in the brain and can distort the neuronal synapses. Lower Aβ levels can be achieved by decreasing BACE1, which may also reverse microglial activation and abnormality in neuronal functions (Coimbra et al., 2018; Yan et al., 1999). Accumulation of Aβ aggregates in the synapses affects synaptic plasticity leading to a decline in memory. BACE1-deficient mice exhibited cognitive impairments significantly. For this reason, BACE1 inhibition is logically beneficial for AD patients (Laird et al., 2005).
A reduction in Aβ deposition caused deletion of BACE1 in adult mice providing us with genetic evidence (Hu et al., 2018) which likely causes AD. Pharmacological inhibition of BACE1 may slow down the Aβ pathology, but to take advantage of this, treatment should be initiated before the spread of Aβ deposition. Therefore, inhibition of BACE1 may regulate to the extent that it prevents the formation of new plaques and delays the progression to AD (Blume et al., 2018; Das & Yan, 2017).
BACE1 is considered a potential target to treat AD, as it is a prime enzyme involved in the Aβ cascade as shown in Figure 2. Intense efforts are being made to find a drug that can inhibit BACE1, but because it plays an important role in various physiological processes, the challenge is to develop selective and specific BACE1 inhibitors (Cole & Vassar, 2007; Rajendran et al., 2008).
For over 20 years, studies have been done to design and develop a BACE1 inhibitor with good selectivity, bioavailability, and blood-brain barrier (BBB) permeability, but clinical trials indicate a high failure rate of many lead drug candidates. Many BACE1 inhibitors have been shelved due to adverse effects, and an inability to reduce AD-related symptoms even though plaque formation is reduced. Nevertheless, BACE1 inhibition is still considered the best target to stop AD, as it is the neck of the disease.
Verubecestat (MK-8931) is the first BACE1 inhibitor (a small molecule) from the Merck pharmaceutical Company with good bioavailability and BBB permeability. The administration of verubecestat to preclinical animals reduced the Aβ oligomers in cerebrospinal fluid (CSF) and the brain with no adverse effects. Phase 1 clinical trials also showed that it is well-tolerated and reduces the plaque concentration in the CSF. In phase 2 clinical trials, the focus was on testing the cognitive properties of the drug. Verubecestat did not exhibit any improvement in the cognitive capacity of AD participants and the clinical trial was terminated in February 2018. Side effects, like rashes, change in color, insomnia, and weight loss were reported by the participants (Egan et al., 2018; Kennedy et al., 2016; Scott et al., 2016; Zwan et al., 2017).
Lanabecestat, an oral BACE1 inhibitor developed by the AstraZeneca Pharmaceutical Company was tested first on primary cortical neurons of mice, guinea pigs, and dogs prior to clinical trials. A half-life of 9 hours means that it has a long-lasting reduction of Aβ oligomers. In 2014, in collaboration with Eli Lilly, phase I studies began, and the results were satisfactory with a good metabolic and tolerability profile in mild AD patients. Similar to Merck’s verubecestat, lanabecestat also decreased the levels of Aβ in CSF in the treated group. In phase 2 clinical trials that were conducted on over 1400 participants, its potency and safety were analyzed by amyloid positron emission tomography (PET) scans and the levels of Aβ in CSF were measured. The expected outcome of the study was an improvement in cognition and a decrease in Aβ levels in CSF. Unfortunately, the trials had to be discontinued as the lanabecestat treatment did not meet the primary endpoints (Cebers et al., 2017; Eketjäll et al., 2016; Sakamoto et al., 2017).
Atabecestat (JNJ-54861911) is another small-molecular BACE1 inhibitor developed by the Shionogi Pharmaceutical Company. This has entered the phase I clinical trial in collaboration with Janssen pharmaceuticals. The administration of atabecestat (5–150 mg) for 14 days showed a considerable reduction in Aβ levels in CSF and plasma. In another trial termed EARLY (NCT02569398) that was begun in 2015, the participants at risk of developing AD or suffering from mild AD were given the drug for 4.5 years with continuous monitoring of cognitive functions. Unfortunately, Janssen had to announce the termination of this trial as observations indicated a rise in the liver enzymes in a few patients. This suggested that it is associated with liver toxicity and the drug was discontinued on May 17, 2018 (Lahiri et al., 2014; Timmers et al., 2018).
Elenbecestat (E2609) was developed first by the Eisai Pharmaceutical Company in December 2014, as a small-molecule BACE1 inhibitor. After initial trials, a ninth phase 1 clinical trial was conducted in 2016, which was satisfactory at a dose of 50 mg in 16 healthy volunteers. In October 2016, a program called MISSION was initiated to examine the outcomes of PET and hippocampal Aβ volume changes. In 2018, Eisai planned for phase 2 clinical trials as it was found to be effective in reducing the brain Aβ levels and cognitive function. However, in 2019, BIOGEN/Eisai announced the termination of MÏSSION trials due to an unfavorable risk-benefit ratio and cognitive improvement was also not significant. However, the only relief was no hepatotoxicity, which has been a feature in clinical trials of other drugs (Imbimbo & Watling, 2019; Panza et al., 2010).
A new small-molecule BACE1 inhibitor CNP520 (umibecestat) from the Novartis Pharmaceutical Company is being developed. In preclinical models, CNP520 has been shown to reduce Aβ generation and penetrate the BBB with a good selectivity toward the target. No phase 1 clinical trial results have been reported, but phase 2 trials were conducted by Amgen Company aimed at delaying the progression or onset of AD. In July 2019, the study ended prematurely, citing cognitive decline and weight loss in the treatment groups. Researchers continued to monitor the cognition capacity and the level of Aβ in the brain after the treatment was stopped, and they found the cognitive decline and the hippocampus volume loss were reversible (Neumann et al., 2018). Regardless of the failures in clinical trials, discovering a BACE1 inhibitor should not be stopped. Inhibition of BACE1 along with other targets could be a potential strategy and is likely to be more effective in AD. Nevertheless, a breakthrough in the drug discovery for BACE1 inhibition happened on June 7, 2021. The first FDA-approved drug for AD treatment is aducanumab developed by the Biogen and Eisai Company under an accelerated approval pathway (Schneider, 2020).
γ-secretase (Presenilin)1
It is an integral intermembrane protein with many subunits forming a protease complex and consists of four individual proteins: PSEN1, PSEN2, APH1, and Nicastrin. γ-secretase and its components are generally synthesized in the endoplasmic reticulum, but for maturation, it requires the coordination of the ER-Golgi circuit (Basi et al., 2010; Wolfe, 2014).
Presenilin mutations were first seen in connection with familial AD, and further research established that PSEN1 along with structurally close PSEN2 are the catalytic components of γ-secretase, which is involved in the cleavage of APP at varying lengths. In AD, APP cleavage by γ-secretase happens at the CTF99 fragment (a fragment of APP after cleavage by BACE1 enzyme) of BACE1, leading to the formation of Aβ plaques. The hypothesized reason behind the action of γ-secretase is after the cleavage of α- or β-secretase into the large ectodomain of the substrate, which prevents binding to γ-secretase. As the final complex in APP processing, γ-secretase might be contributing to the levels of the Aβ and Notch signaling pathway as shown in Figure 3. Notch receptor is responsible for cell differentiation events in all multicellular organisms, in embryogenesis and adulthood (Basi et al., 2010; Bavishi et al., 2015).
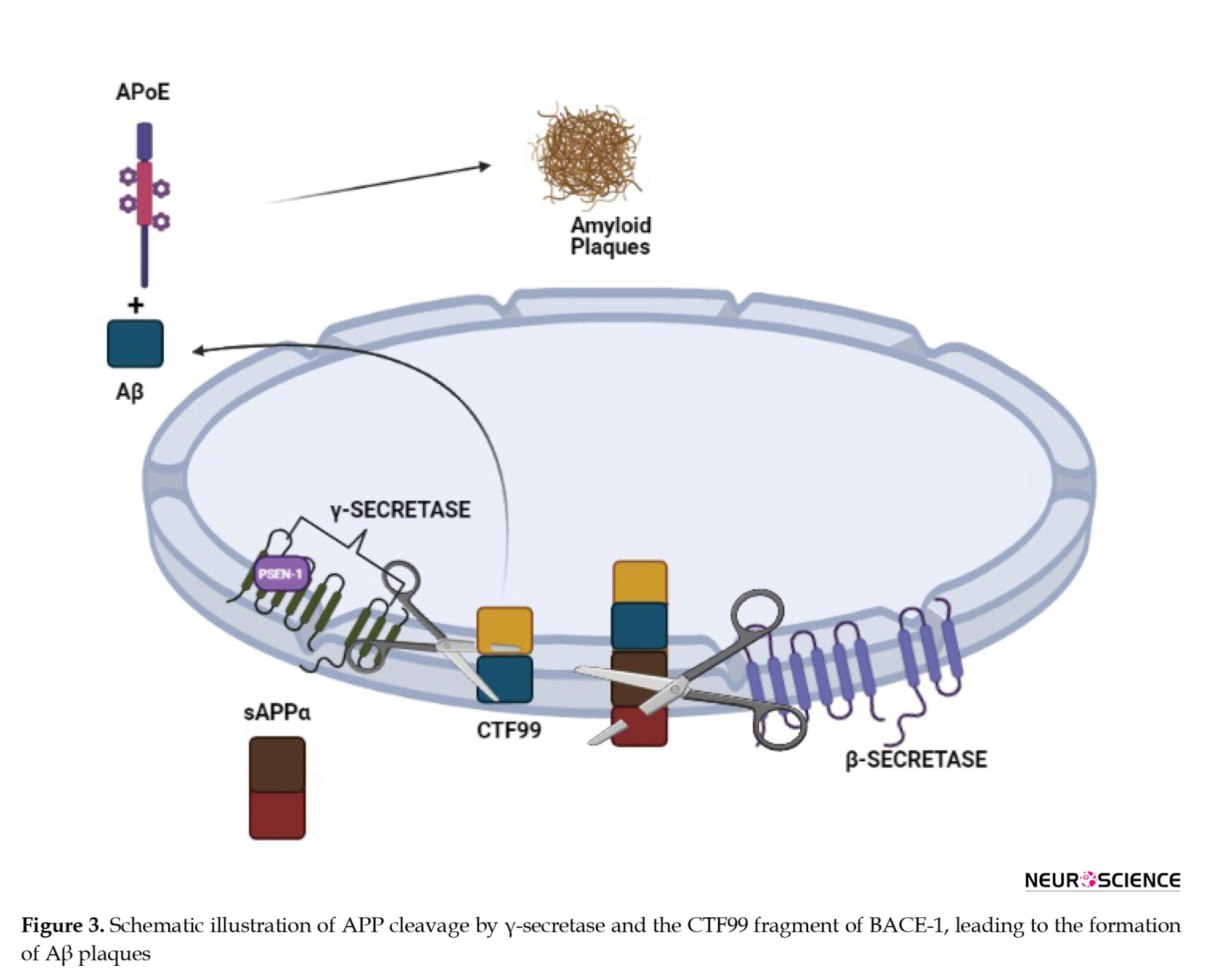
The inhibition of γ-secretase presents an obvious door to block the production of Aβ (De Strooper, 2007; Wolfe, 2014). The first reported γ-secretase inhibitors (GSIs) were peptide aldehyde type calpain and proteasome inhibitors. Despite their less potency and lack of selectivity, these are said to be the answers for GSIs (Wolfe, 2012).
The complete inhibition of γ-secretase seemed a good approach. However, it was found that it has an extensive biological role and cleaves multiple proteins to yield physiologically essential products. The total inhibition of γ-secretase has resulted in severe adverse effects in vivo. This has been evident from the clinical trial conducted by Eli Lilly for the γ-secretase inhibitor called semagacestat. Patients experienced severe side effects, which led to the discontinuation of the clinical trial in 2010 (Kounnas et al., 2010; Schiering et al., 2016).
A substantial amount of research is being done for the discovery and development of small-molecule inhibitors for presenilin-containing γ-secretase complex as a potential therapy for AD (Kounnas et al., 2010).
The future focus will be developing an inhibitor that can reduce the Aβ levels without being involved in the Notch signaling pathway and discovering γ-secretase modulators, which can shift the Aβ1-42 to less pathogenic forms. Pharmacological inhibition of γ-secretase is a well-documented target for lowering plasma Aβ levels and has a greater impact on the pathology of AD (De Strooper et al., 2012; Wolfe, 2014).
PF-3084014, as a γ-secretase inhibitor, was in development in Pfizer but has to be abandoned due to the lack of data on its effect on cognition and the amount of Aβ plaques in animal models of AD (Panza et al., 2010).
GDI-953 a potent γ-secretase inhibitor developed by Wyeth has been shown to inhibit Aβ production at nanomolar concentrations in preclinical animals but in phase 2 clinical trials, it did not show much effect on the AD patients. The dose administered was not sufficient to clear the plaques or improve the cognition capacity of AD patients (Martone et al., 2009).
E-2012, a novel γ-secretase inhibitor reduces Aβ levels by modulating γ-secretase without interfering with the Notch processing. This drug was developed by Eisai in collaboration with torrey-pines therapeutics. In 2006, the drug candidate entered into phase 1 clinical but was terminated because of the side effects observed in preclinical animals after 13 weeks. The drug is now back and the clinical trial is under process and is in phase 1 (Panza et al., 2010).
FRM-36143 is another novel γ-secretase inhibitor that reverses the PSEN mutations without interfering with the Notch signaling pathway in the preclinical studies. Thus, FRM-36143 is a potential candidate for further research to take into the clinical sector (Blain et al., 2016).
As γ-secretases play multiple roles, targeting PSEN1 mutations specifically will be the best way to minimize and prevent disturbances in the physiological functions of the body.
Cdk5
Two decades have passed since the discovery of CDK. Cdk5 belongs to the family of cyclin-dependent kinases, which are necessary for brain development during embryogenesis and essential for various neuronal functions related to cognition and memory in adults. Cdk5 is mainly found to be active in post-mitotic neurons (Dhavan & Tsai, 2001).
In a normal brain, the activity of Cdk5 is highly regulated by specific cyclin-related molecules p35 and p39. Among the two, p35 is the most studied activator protein of Cdk5. Regulated Cdk5 maintains synaptic plasticity, the survival of neurons, and cognition. When the activity of Cdk5 is deregulated, it promotes oxidative stress and mitochondrial dysfunction (Shukla et al., 2012; Wang et al., 2015; Wilkaniec et al., 2018).
In case of stress, there is an increase in the level of calcium in the cytoplasm, leading to proteolytic cleavage by calpain. Calpain belongs to the family of calcium-dependent cysteine proteases that are involved in cognition and cell development, motility, apoptosis, and differentiation. Calpain has two forms associated with it m-calpain and μ-calpain. They require only milli molar and micro molar concentrations of calcium for their activation. Calpain cleaves p35 and p39 resulting in p25 and p29 as cleaved fragments. This p25 can activate Cdk5 leading to a higher and active Cdk5/p25 stable complex. This causes further Aβ formation, mitochondrial dysfunction, and other pathological events leading to degenerated neurons and apoptosis as represented in Figure 4 (Cruz & Tsai, 2004; Liu et al., 2003). In AD, plaques are formed due to the deposition of Aβ aggregates extracellularly. Over-expression of Cdk5 promotes further Aβ deposition resulting in neurotoxicity, activation of kinases, and further neurofibrillary tangles (NFT) formation (Liu et al., 2003; Reinhardt et al., 2019; Taniguchi et al., 2001).
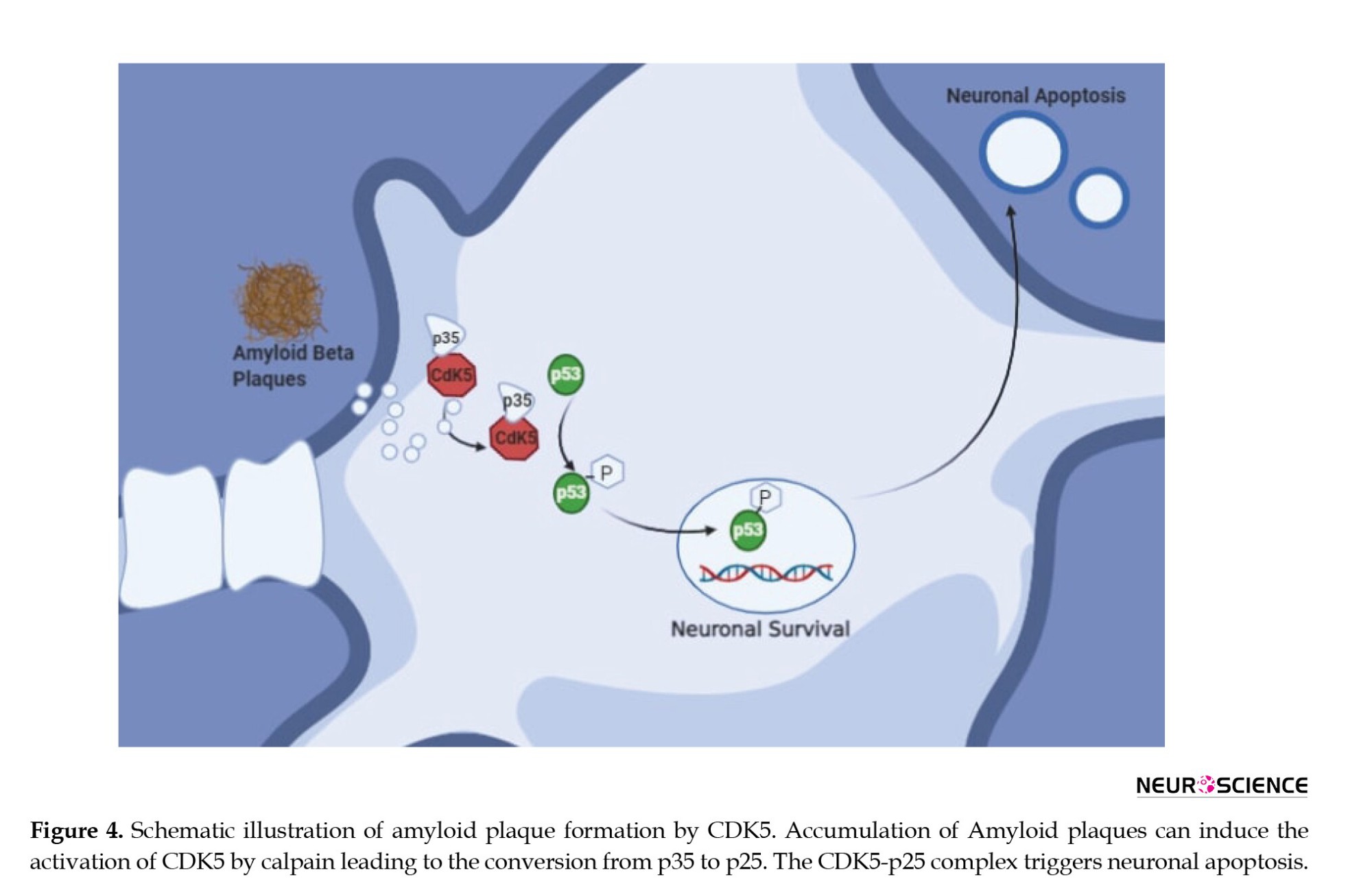
One study showed that upon neurotoxicity by glutamate or Aβ, neurons up-regulated Cdk5 activity, but upon treatment with known Cdk5 inhibitors, cells showed a reduction of hyperactive Cdk5. This strongly indicates that hyperactivation of Cdk5 may be involved with phosphorylation of APP leading to excessive Aβ formation (Cruz & Tsai, 2004; Fischer et al., 2002; Shah & Lahiri, 2014). An increase in Cdk5 activity of p25 is observed in the post-mortem brains of AD patients making it a therapeutic target for AD.
In research related to Cdk5 inhibition, two inhibitors have been tested: Cdk5 inhibitor (roscovitine) (Huber & O’Day, 2012; Menn et al., 2010) and calpain inhibitor (MDL28170) for AD. Roscovitine is in phase 2 clinical trials for the treatment of lung and nasopharyngeal cancer. Cdk5 deregulation is related to melanoma and neurodegenerative diseases, suggesting that roscovitine can be further investigated for AD. Roscovitine exhibits neuroprotective activity in vitro and in vivo (Dhavan & Tsai, 2001; Khalil et al., 2015; Shah & Lahiri, 2014).
Studies on AD mouse models suggest that the inhibition of Cdk5 activity genetically or pharmacologically will prevent synaptic dysfunction and neuronal death (Seo et al., 2017). Trials with Cdk5 inhibitors in the clinical sector have not been promising due to side effects or off-target events (Cicenas et al., 2015).
AK275 is the first exogenous calpain inhibitor and was mostly preferred because of its selectivity, membrane permeability, solubility, and in vitro efficiency. AK275 is considered to be a neuroprotectant for the treatment of focal brain ischemia, indicating its potential for treating AD (Yildiz-Unal et al., 2015). There are other inhibitors that researchers are working on to improve their stability, potency, and efficiency with improved pharmacological profiles (Bartus et al., 1994; Wilkaniec et al., 2016).
In a recent study on mouse models of AD, anti-diabetic drugs troglitazone and pioglitazone belonging to the class of thiazolidinedones (TZD) have been shown to inhibit Cdk5 kinase activity and reduce synaptic deficits (Cho et al., 2013). However, the use of TZDs is more than usually related to edema, weight gain, and heart failure (Rizos et al., 2009), which raises a concern.
In another study on APP/PS1 mice, in which Cdk5 was hyperactivated in the hippocampus, an anti-diabetic drug metformin was administered. The reports showed that after chronic administration of metformin, the cleavage of p35 into p25 was prevented and in turn, inhibited Cdk5, which was assessed by a combination of pharmacological and molecular techniques. Also, the reports have shown that chronic administration of metformin restored impaired synaptic plasticity and improved cognitive function in mice (Wang et al., 2020). Collectively, this information suggests a captivating note that anti-diabetic drugs can be of use as drugs to treat neurodegenerative diseases.
In the end, drug candidates that can reduce the hyperactivity of Cdk5 may be regarded as a highly potential candidate to decrease the production of Aβ (Fischer et al., 2002).
Dual-specificity tyrosine-regulated kinases (DYRK1A)
DYRK1A is a dual specific tyrosine-phosphorylated regulated kinase enzyme that belongs to the DYRK family and is one among five protein kinases that direct signals in the development of the nervous system (Ferrer et al., 2005; Yang et al., 2001). DYRK1A is extensively found in the cerebellum, olfactory bulb region, and hippocampus, and is also present in the Down syndrome region on chromosome 21, and controls brain growth through neuronal proliferation and neurogenesis. DYRK1A is involved in the formation of Aβ plaques and NFT, which has been evident from the reports of higher concentrations of DYRK1A in the brains of AD patients (Souchet et al., 2019; Stotani et al., 2016). Although DYRK1 plays a role in the formation of plaques, it is essential for the brain during embryonic development in the early stages, where DYRK1 is involved in signaling pathways related to cell growth and proliferation, separation of cells into mature neurons, as well as in the formation of dendritic spines, which are important for the transmission of impulses. However, in a mature brain, DYRK1A can become aggressive and may initiate AD pathology (Galceran et al., 2003). The dysfunction of DYRK1A is a prominent feature of Down syndrome and patients with this disorder are highly susceptible to developing AD early in life (Velazquez et al., 2019).
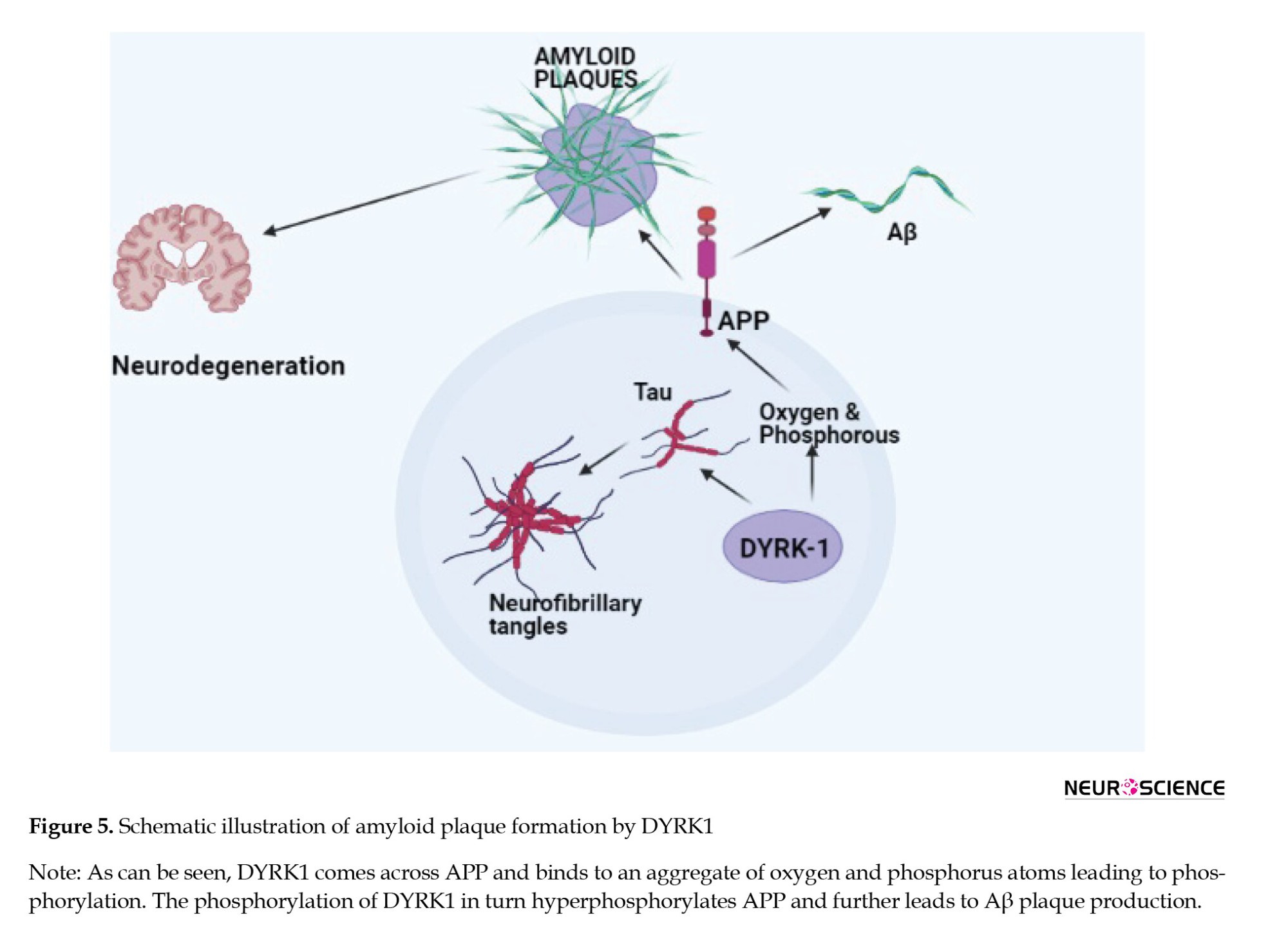
When DYRK1 comes across APP, it binds to an aggregate of oxygen and phosphorus atoms leading to phosphorylation. High phosphorylation of these proteins leads to harmful effects in the brain. This hyperphosphorylation of APP leads to the formation of Aβ plaques as shown in Figure 5. Experimental evidence shows that the over-expression of DYRK1A contributes to enhanced β-amyloidosis and neurofibrillary degeneration. The over-expression of DYRK1A has been shown to increase the chances of proteolytic cleavage of APP and hence, an increased production of Aβ peptides. Besides, DYRK1A phosphorylates presenilin 1 (PS 1) and thereby increases γ-secretase activity causing more Aβ production (Souchet et al., 2019; Yang et al., 2001).
A drug repurposing study of CX-4945, a CK-2 inhibitor undergoing clinical trials for various cancers, shows that it inhibits DYRK1A-related AD pathology in mouse models (Chon et al., 2015). The inhibition of DYRK1A in chronic conditions has reversed memory problems in 3x-Tg AD mice. The results obtained were found to be associated with Aβ and tau pathology. It showed that the inhibition of DYRK1A reduced the Aβ plaque formation by inhibiting APP cleavage and also reduced insoluble tau phosphorylation (Branca et al., 2017; Pathak et al., 2018).
This suggests that drugs that can inhibit DYRK1A could be a possible therapeutic choice for AD. Knowledge of the mechanisms initiated by DYRK1A over-expression will get us closer to comprehending AD and developing effective therapies.
Some DYRK1A inhibitors have been tested in vitro but only a few have shown the potential to be clinical drug candidates. Harmine is an extensively studied DYRK1A inhibitor in preclinical research but because of its selectivity toward kinases, is not suitable for clinical development (Melchior et al., 2019).
ManRos Therapeutics is developing a small-molecule DYRK1A inhibitor named leucettine L41 based on the analog of leucettamine B derived from marine sponges (Burgy et al., 2013).
Samumed is developing a DYRK1A inhibitor named SM07883, which has been shown to protect against the hyperphosphorylation of tau and is currently under test in a phase 1 clinical trial (Melchior et al., 2019).
KVN93, a molecule that has considerably reduced DYRK1A kinase activity in vitro, is speculated to be a potential novel DYRK1A inhibitor that can improve cognitive deficits and regulate Aβ pathology (Lee et al., 2020).
Using the natural product epigallocatechin-3-gallate, the inhibition of excess DYRK1A activity and promoted improved cognitive functions were observed. However, these drugs are non-selective to the target, and possessing numerous off-target reactions has raised questions on their utility (Feki & Hibaoui, 2018).
Preclinical research shows that DYRK1A inhibitors are effective in slowing down the onset of AD in people suffering from Down’s syndrome. Despite many efforts to develop DYRK1A selective inhibitors, only a few of them have crossed the preclinical stage and their clinical manifestation remains to be tested further. The safety parameters are the primary concern for DYRK1A inhibitors and this depends on their kinase selectivity and potency toward the target.
Apolipoprotein E (APOE)
APOE is a gene present on chromosome 19 and makes a protein APOE that helps in the metabolism of fats in the body (Liu et al., 2013; Yamazaki et al., 2019). APOE has two regions an N-terminal, which is the receptor-binding region, and a C-terminal domain, which is a fat-binding region. The APOE gene in humans is classified into three polymorphic alleles (E2, E3, and E4) with a prevalence rate of 8.4%, 77.9%, and 13.7%, around the world. However, the frequency of the E4 allele is remarkably more up to 40% in AD patients. APOE consists of 299 amino acids with a molecular weight of 34 kDa. The number of amino acid residues differentiates the three APOE isoforms (Morris et al., 2010). An in vitro study in 1996 showed that APOE is involved in Aβ formation and the APOE4 allele is considered to be the driving factor over the E2 or E3 isoform (Adalbert et al., 2007). APOE is mainly found in the liver and brain and helps in transporting lipids among cells or tissues by binding to lipoprotein via the APOE4 receptor in the liver. ApoE4 slows down the rate of transportation of cholesterol from the blood, which may lead to coronary heart disease. In the brain, APOE is mainly secreted in the astrocytes. Various experiments have indicated that APOE isoforms can efflux cholesterol. APOE2 and APOE3 have a better efficiency than APOE4 in reducing the cholesterol levels. Cholesterol is needed for different functions of neurons, which include synaptic plasticity and maintaining dendritic spine density. The less ability of APOE4 in lipid metabolism affects the condition of a neuron leading to neuronal death as seen in Figure 6. Carriers of APOE4 tend to have enhanced pathology related to AD before clinical symptoms become evident. APOE4 also presents the pathogenesis of AD by independent Aβ mechanisms, which include changes in the synaptic plasticity, lipid homeostasis, and neuronal inflammation. The precise pathophysiology between APOE isoforms and Aβ metabolism is still unclear. Current studies have found out that APOE4 could alter the production, aggregation, and clearance of Aβ (Namba & Ikeda, 1991; Safieh et al., 2019). Furthermore, APOE4 carriers have lower levels of CSF Aβ42 and show higher PiB and PET binding than non-carriers (Liu et al., 2013). Moreover, APOE4 is believed to enhance Aβ production by affecting the activity of γ-secretase (Adalbert et al., 2007; Liu et al., 2017).
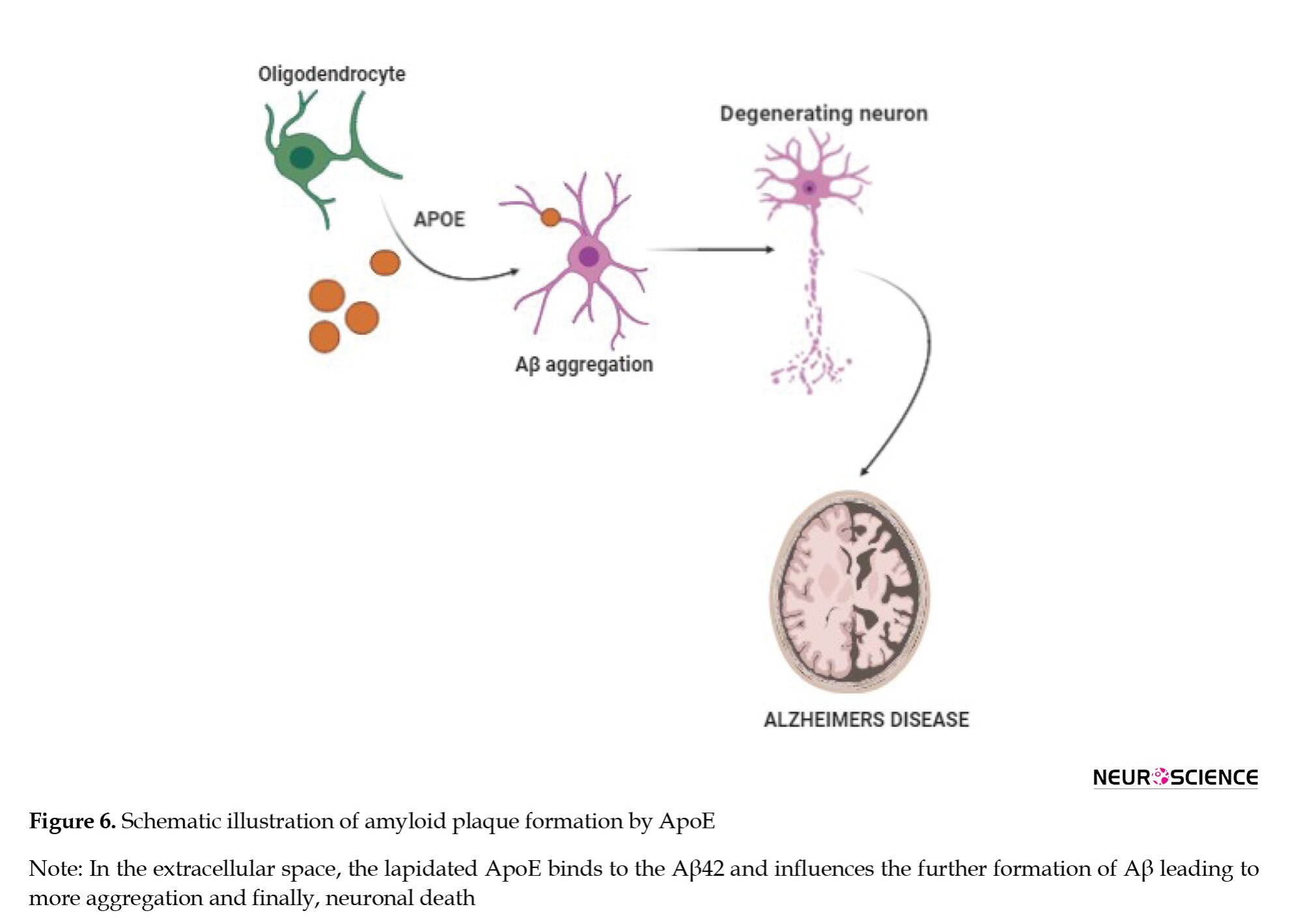
APOE2 is considered to be a protective allele against AD. APOE2 could delay AD by an average of 7.5-8.5 years. Scientists have developed an Adeno-associated virus (AAV) type of particle that was able to reduce the Aβ load and bring back the cholesterol to normal levels in transgenic mice. Bexarotene as a retinoid x receptor (RXR) reversed both brain pathologies and cognitive dysfunctions that are driven by APOE4 and also induced compensation for the lipid deficiency of APOE4. This suggests that RXR agonists may be a way to counteract the pathological effects of APOE4 leading to AD (Boehm-Cagan & Michaelson, 2014).
A recent in silico study reported that epicatechin-gallate has a binding affinity toward APOE4 and can be a potential lead compound to inhibit the progression of AD (Bano et al., 2019). Thus, elucidation of the pathogenic link between APOE4 and memory functions is a considerable challenge. Inhibitors selectively targeting APOE4 or increasing the expression of APOE2 could bring assistance in the way of drug development for AD. Another strategy that can be implemented is to edit the gene from the E4 variant to the E3 variant.
Casein kinase 1 (CK1):
Exactly two decades ago, the regulation of casein kinase in AD was reported. Casein kinase 1 belongs to the family of protein kinases, which are mainly involved in maintaining circadian rhythms. Until now, seven different isoforms of CK1 have been characterized, of which CK1-δ and CK1ε are expressed in the brain (Rodrigues & Silva, 2017).
CK1-δ and CK1ε play an important role in maintaining homeostasis of the circadian rhythm (Figure 7).
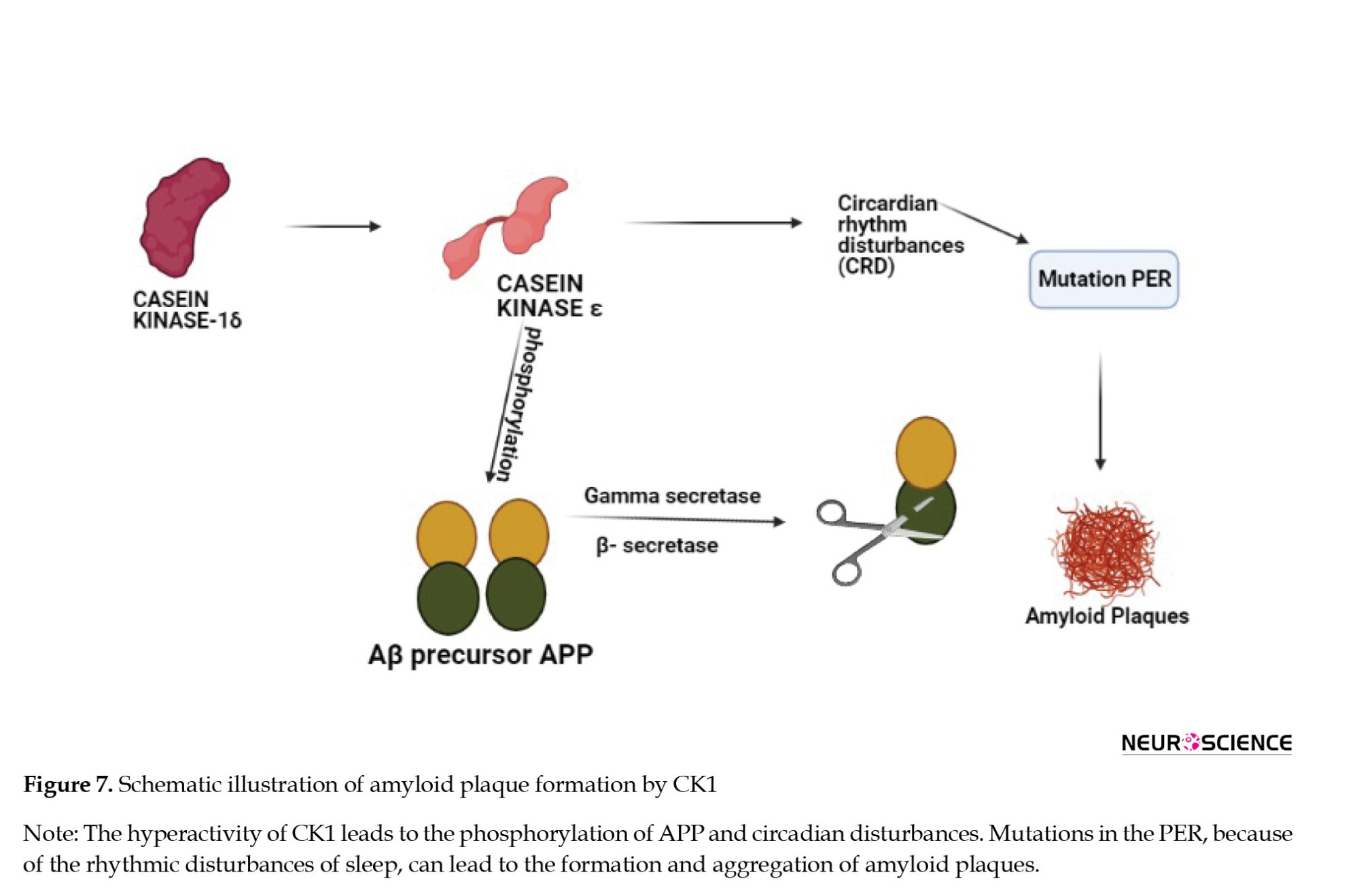
In specific genetic ablation, CK1-δ changes the period of circadian rhythm and CK1ε may result in arrhythmicity. This is one of the reasons subjects with AD go to sleep later than the rest of the population. Disruption of the clock by CK1 hyperactivity and mutations in the period gene PER may be a causative factor in the pathogenesis that underlies AD as described in Figure 8. An in vitro study demonstrated that CK1ε can phosphorylate the Aβ precursor APP and both β- and γ-secretases that can cleave APP (Chauhan et al., 1993; Yasojima et al., 2000).
There is an involvement of CK1 activity in the regulation of APP cleavage (Flajolet et al., 2007). A higher association of CK1 is seen in neurodegenerative lesions in the final stages of neuronal death in the brain, which is the same with AD. The key enzymes that are involved in the cleavage of APP (β-secretase and γ-secretase) are also related to CK1 as a target to inhibit plaque formation. The brains of AD patients have 30 times more CK1 than that of a normal human brain (Adler et al., 2019; Chauhan et al., 1993).
A research study (2019) done on APP-PS1 mice showed that the inhibition of CK1 improves cognitive function and also reduces the load of Aβ in the brain. Another study reported that Aβ stimulates casein kinases along with other protein kinases, which may then initiate tau hyperphosphorylation leading to neurofibrillary tangles and ultimately neuronal death (Manakadan et al., 2015; Sundaram et al., 2019). Further characterization and identification of new inhibitors of CK1 that can regulate AD-regulated circadian shifts can be focussed as it is essential for the clearance of Aβ.
A small-molecule inhibitor -PF-670462- of CK1δ/ε is found to alter rhythmicity, improve cognitive functions, and reduce the Aβ load in APP-PS1 mice (Sundaram et al., 2019).
PF-05251749, a CK1δ and ε inhibitor, initially developed by Pfizer Company for AD, was tested for phase 1 safety and tolerability following preclinical studies considering circadian rhythm as a major parameter for assessment. No further development has been updated since October 2018; however, Biogen acquired the molecule for further studies in the AD population in January 2020 (Benn & Dawson, 2020). Future research should concentrate on early intervention to reduce CK1ε/δ by understanding the prophylactic potential.
Nod-like receptor protein 3 (NLRP3)
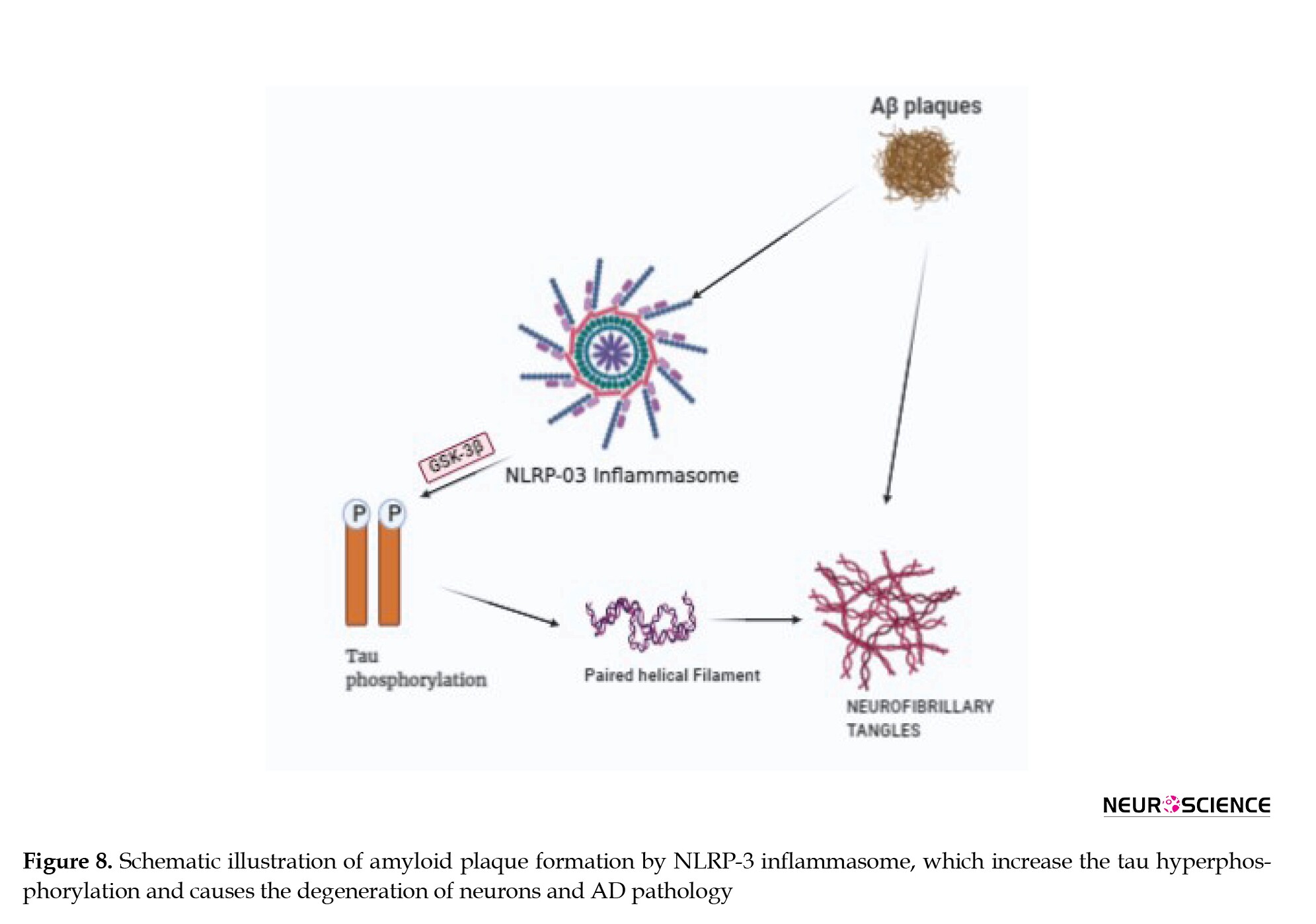
NLRP3 is an intracellular oligomeric multi-protein complex that is being extensively studied for neurodegenerative and immune system-related diseases (Song et al., 2017). NLRP3 is also called an inflammasome that belongs to the family of nod-like receptor (NLR) and is present in astrocytes and microglia in the CNS and elicits an immune response against damaged signals. The NLRP3 protein complex consists of the sensor protein NLRP3, the adaptor protein, a caspase activating and recruiting domain, and pro-caspase-1. The assembly of this complex and its activation leads to the separation of procaspase-1 into active caspase-1. The activated caspase-1 in turn cleaves and activates proinflammatory cytokines. In AD, the aggregation of misfolded proteins or Aβ plaques triggers NLRP3 can act as a platform for the activation of caspase-1 that initiates an inflammatory response, which triggers the pro-inflammatory cytokines IL-1β and IL-18 that are released into the extracellular space. These promote various inflammatory and immune diseases and mediate neuronal death (Halle et al., 2008; Yang et al., 2018; Yang et al., 2019, Tan et al., 2013). The inflammasome constituents, such as NLRP1, NLRP3, ASC, and Caspase-1 along with the downstream effectors, like IL-1β and IL-18 are up-regulated at both mRNA and protein levels in AD patients Figure 8 describes the pathway of Aβ plaque formation by NLRP3.
A study conducted in 2013 assessed the amount of cleaved caspase-1 in AD patients and found that its levels were more compared to the controls. The same case was mirrored in aged APP/PS1 transgenic mice (Yin et al., 2018).
A study of a rationally designed NLRP3 inflammasome inhibitor (JC-124) in Transgenic CRND8 APP mice showed that upon treatment with (JC-124), the levels of Aβ deposition in the brains of CRND8 mice were reduced along with reduced APP cleavage (Heneka et al., 2013). This suggests that discovering agents that can control the activation of NLRP3 might be a potential target to tackle neuroinflammation and plaque formation.
Since NLRP3 inflammasome activation is a multistep process, inhibiting the activation of NLRP3 can be done by different approaches: Subduing molecules that initiate the NLRP3 complex formation, suppressing the upstream signals by inhibiting the NLRP3 directly or indirectly depending on the molecule targeted and inhibiting caspase-1 cleavage. Several molecules have been shown to inhibit NLRP activation and are said to have been validated in animal models. An in vivo study described that a potent small-molecule inhibitor MCC950 blocked NLRP3 activation selectively at nanomolar concentration (Coll et al., 2015). A recent study on MCC950 also demonstrated that it can block nigericin-induced activation of NLRP3 inflammasome by inhibiting efflux of chloride that acts as an upstream pathway for NLRP3 activation (Zahid et al., 2019). Cystic fibrosis transmembrane conductance regulator (CFTR) channel inhibitor C172 can also inhibit NLRP3 activation. Another report on C172-CY09 confirms that it selectively binds to NLRP3 inflammasome but not to any other and inhibits its ATPase activity. The ATPase activity of NLRP3 is important for the activation of NLRP3 and its oligomerization (Jiang et al., 2017).
Most predominantly, CY-09 showed an inhibitory effect on NLRP3 and good pharmacokinetic properties in terms of tolerability and bioavailability in a mouse model related to NLRP3-related disease. CY-09 is the first compound identified that was specifically able to inhibit NLRP3 both in vitro and in vivo. However, additional studies are needed for its selectivity towards NLRP3 (Jiang et al., 2017; Lamkanfi & Dixit, 2017).
OLT1177, which was identified as a candidate for the treatment of degenerative arthritis, has successfully cleared the phase I clinical trial, and now is in the phase 2 clinical trial for the treatment of acute gouty arthritis. Marchetti et al. described that OLT1177 possesses anti-inflammatory action and can inhibit NLRP3 inflammasome activation. Like CY-09, OLT1177 also directly binds to NLRP3 and inhibits ATPase activity (Marchetti et al., 2018; Toldo et al., 2019).
To date, many molecules have been identified and investigated as NLRP3 inflammasome inhibitors in mouse models, but only a few have clinical value. However, a new study identified that the anti-allergic drug, tranilast can be used to treat inflammatory diseases. Huang et al. reported that tranilast can inhibit NLRP3 specifically. Like the other specific inhibitors, tranilast does not interfere with the signaling pathways of NLRP3 inflammasome. Tranilast can also inhibit NLRP3 inflammasome activation in an ATPase-independent manner. Further, in vivo studies have shown that tranilast has a preventive effect on mouse models related to NLRP3-related diseases. The safety profile needs to be investigated further (Feng et al., 2020; Huang et al., 2018).
A widely used OTC herbal medicine for the treatment of inflammatory diseases, Rabdosia Rubescens, has an active constituent, oridonin, which is reported to exhibit anti-inflammatory effects (Kuo et al., 2014). Previous reports have mentioned that oridonin can suppress inflammatory mediators and exhibit therapeutic effects on neuroinflammation. Studies have suggested that oridonin can specifically inhibit NLRP3 inflammasome activation (Kuo et al., 2014; Wang et al., 2019; Wang et al., 2015; Yang et al., 2019). It is used in clinical practices for different diseases but studies need to be further done for its therapeutic potential in NLRP3 inhibition associated with AD.
Matrix metallopeptidases (MMP)
Matrix metallopeptidases are also known as matrixins or matrix metalloproteinases and are calcium-dependent proteases belonging to the metzincin superfamily. The MMP family consists of other 28 MMPs, which are enzymes capable of degrading and processing many proteins in the extracellular matrix. MMPs are further divided into six main subgroups, of which gelatinase type (MMP-2 and 9) and membrane-type MMPs (MMP-1, 2, 3, 4, 5, and 6) play a role in AD and other neurological diseases (Wang et al., 2014; Yin et al., 2006). MMPs play an important role in cytokine inactivation responsible for inflammation, and physiological processes, such as neurogenesis and angiogenesis. MMPS is generally expressed in neurons but is also found in astrocytes and microglia. Astrocytes have been recently considered mediators in the degradation of Aβ. Evidence suggests that MMP-2, MMP-3, and MMP-9 are important players in AD compared to the other MMPs (Brkic et al., 2015; Duits et al., 2015; Yin et al., 2006; Yoshiyama et al., 2000)
MMP-2 is the major MMP that is directly linked to Aβ in the brain and the dysfunction of this enzyme exerts influence on the processing of Aβ(1-40/42) in vitro and in vivo, as deletion of MMP-2 leads to a greater Aβ accumulation rather than MMP-9 knockout. The proteolytic activity of MMP-2 and MMP-9 in clinically diagnosed AD patients showed higher activity of MMP-9 but not MMP-2 when compared to healthy brains suggesting that MMP-2 may have a protective role in AD (Yin et al., 2006). Expression MMP-9 is seen in cytoplasm, NFT, senile plaques, hippocampus, and cerebral cortex in AD patients. A post-mortem report of human frontal and parietal cortical tissues obtained from clinically diagnosed AD patients has shown a higher activity of MMP-9 rather than MMP-2 compared to healthy brain samples. A recent study also found that astrocytes present near the Aβ plaques have shown an increased expression of MMP-9 in aged APP/PS1 mice (Brkic et al., 2015; Fragkouli et al., 2014). From these reports, a piece of conclusive evidence can be drawn that MMP-9 is over-expressed and is found to be higher in the plasma of people with AD. As AD is a complex disorder with many agents playing a role, targeting more than one MMP with pan-specific inhibitors or combination of MMP with another target of AD can also be explored (Lorenzl et al., 2003).
Minocycline, an anti-inflammatory drug that is used for multiple sclerosis and Huntington’s disease, was tested for its effect on AD. It showed good tolerability and pharmacokinetic profile in vitro and in vivo. However, during a randomized clinical trial conducted on AD patients, it did not show any improvement in the cognitive capacity nor did it slow down the progression of AD. The negativity is explained because of the poor dose that has been tested in animals. This can be further explored for AD by altering the doses in the preclinical studies and thereby testing in humans considering that minocycline has a good penetration of BBB (Howard et al., 2020; Rosenberg et al., 2015; Yong et al., 2007).
MT-5-MMP is a protease that has come into the limelight as a potential target related to Aβ plaques in AD. A study on a 5xFAD mouse model of AD showed that MT5-MMP deficiency reduced Aβ levels in the cortex and hippocampus suggesting that inhibitors targeting MMP-5 specifically can be explored for AD (Baranger et al., 2016).
ZHAWOC7726, a TIMP peptidomimetic protein, has a good selectivity toward MMP-9, MMP-12, and MMP-13 for the treatment of cancer. This molecule could be further studied as it acts on different MMPs associated with AD (Gall et al., 2019).
Neprilysin (NEP)
Neprilysin, also called membrane metalloendopeptidase (MME), is an enzyme encoded by the MME gene in humans and cleaves peptides at the amino side of hydrophobic residues (Vodovar et al., 2015). It is found in a variety of tissues but more prominently is expressed in the kidneys and to a lesser amount in the brain and is an enzyme that controls Aβ degradation. NEP has a higher affinity toward the Aβ than other neuropeptides. Figure 9 shows the role of neprilysin in the degradation of Aβ plaques (Marr & Hafez, 2014; Webster et al., 2014).
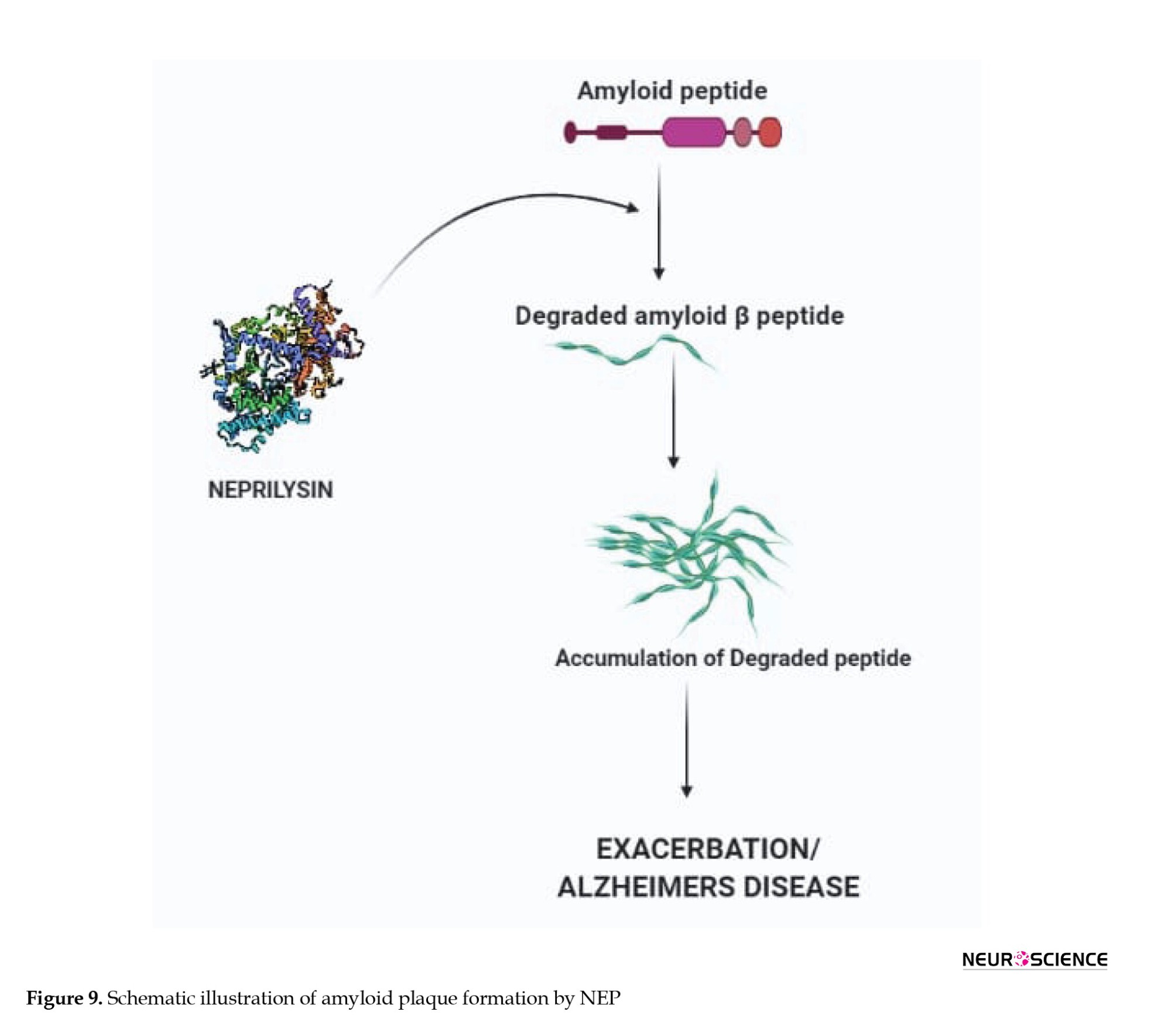
During the early stages of AD, NEP is inactivated and down-regulated. Early studies have suggested that maintaining NEP levels in the brain could be a potential therapeutic strategy to slow the progression of AD (El-Amouri et al., 2008; Huang et al., 2006; Madani et al., 2006). The importance of NEP in Aβ regulation was demonstrated by using NEP knockout experiments. Elevated levels of Aβ species were seen in the hippocampus and brainstem when mice deficient for the NEP2 gene were used. Increased Aβ levels were observed in NEP2 knockout mice mated with APP Tg mice (El-Amouri et al., 2008; Yasojima et al., 2000).
The discovery of endopeptidases, such as NEP and NEP2 has opened up doors for viral-mediated gene therapy. While NEP has been a prominent target, NEP2 also can be considered selectively for Aβ clearance. A study has reported that the destruction of NEP has increased the levels of Aβ in the brain of mice (Huang et al., 2006; Madani et al., 2006; Maruyama et al., 2005).
Taking into account that NEP expression and activity are reduced in AD, the idea of increasing the NEP up-regulation might be beneficial. Saito et al. reported that somatostatin, a neuropeptide, and a NEP substrate can up-regulate NEP activity by regulatory the feedback cycle. Activation of somatostatin receptor subtype 4 increases the activity of NEP in the cortical regions suggesting that somatostatin can be considered an interesting target for increasing NEP levels (Bavishi et al., 2015; Eckman & Eckman, 2005). Hence, effective strategies and studying the pathways to increase the levels of NEP expression and activity may give us new opportunities for therapeutic interventions in AD as NEP inhibition has been a successful therapeutic candidate in heart failure with less ejection fraction (Pavo et al., 2020).
Dopamine 2 receptor (D2R)
Dopamine is a neurotransmitter and is generally expressed in the limbic system and cortex, which is related to mood and emotional stability (Ambrée et al., 2009; Kemppainen et al., 2003). Dopamine generally acts through five different receptors (D1, D2, D3, D4, and D5). Cortical functions are influenced by the number of dopamine (D2) receptors available. AD is associated with deficits in several neurotransmitters but cholinergic and dopamine systems are the most investigated in neuronal research. The levels of dopamine were found to be higher in AD patients than in controls (Martorana & Koch, 2014; Reeves et al., 2017).
Dopamine-containing neurons are mainly located in the midbrain. Dopamine plays an important role in synaptic transmission. Being a neuromodulator, there are chances of dopamine affecting the neurotransmitter release and membrane excitability of the pre and post-synaptic cells (Nobili et al., 2017). Dopamine can modulate the activity of cholinergic release from the neurons located in the basal forebrain. The dysfunction of dopamine is being seen as a new player in the pathogenesis of AD (Liu et al., 2016).
A 1986 report showed that the administration of levodopa restored cholinergic neurotransmission (Rinne et al., 1986). Dopamine regulates synaptic plasticity and also can maintain cognitive functions. Maintaining dopamine levels may reduce plaque formation by increasing synaptic plasticity and reducing oxidative stress (Cross et al., 1981; Martorana & Koch, 2014; Pan et al., 2019).
A recent study showed a link between the ventral tegmental area (VTA) and other parts of the brain. They analyzed the subjects by using 3-Tesla MRI technology. The reports indicated that there is a relationship between size and the functionality of VTA, the size of the hippocampus, and learning ability. The smaller size of VTA indicated that only a lesser amount of dopamine goes to the hippocampus and reduced memory performance. They concluded that this might be the reason for AD patients have memory loss, suggesting agents that can increase the dopamine levels in the body can reduce plaque formation and the progression of AD (Pan et al., 2019). Thus, maintaining the dopamine levels in the brain can at least delay the progression of AD by stabilizing the signaling pathway and reducing neuronal degradation.
Triggering receptor expressed on myeloid cells 2 (TREM2)
In the CNS, TREM2 is expressed mainly in microglia and is involved in the production of proinflammatory cytokines and controls neuronal inflammatory events as depicted in Figure 10 (Gratuze et al., 2018; Karanfilian et al., 2020). Being a transmembrane receptor, the involvement of immune and inflammatory pathways supports the fact that it has a role in AD. TREM2 is necessary for maintaining the microglial progression to fully mature disease-associated microglia (DAM). The expression of TREM2 is associated with plaque formation. Microglia respond to Aβ accumulation and become DAM (Jay et al., 2017).

Microglia have been shown to surround and limit Aβ plaques in the AD brain in the absence of microglial scavenging receptors and resulted in decreased clearance of Aβ by microglia (Carmona et al., 2018; Jay et al., 2017; Zheng et al., 2018). So far, 46 variants of TREM2 have been worked upon about AD. A rare genetic variant p. Arg47His (rs 75932628) is said to have an increased risk of developing AD in the European and American populations. However, the association of the same has not been prominent in Asian countries, suggesting that TREM2 may play a population-specific role (Jay et al., 2017).
Several in vitro and in vivo studies have been done concerning TREM2 and Aβ burden. In vitro studies have shown that TREM2 is strongly involved in Aβ40 and Aβ42 uptake by the microglial cells and plaque formation. In vivo studies conducted on different mouse models suggest an age-dependent effect on Aβ deposition (Cheng et al., 2016).
TREM2 may result in greater Aβ deposition in the early stages of the disease and then becomes beneficial in the end stages by interfering with inflammatory signaling and maintaining the ability of microglia to recover the injured neuron (Carmona et al., 2018). A full understanding of the dual role of TREM2 in neurodegenerative disease, especially in AD is important. Addressing questions on whether altered phagocytosis of TREM2 is the reason for the accumulation of Aβ plaques or is it the interaction of TREM2 with other targets of AD can be a viable therapeutic strategy.
PY134 developed by Pionyr’s clinical programs for specifically targeting TREM2 in cancer is in phase 1 clinical trial. As the molecule can bind to TREM2 specifically, further studies can be done to explore its efficacy in AD (Tang et al., 2019).
Activating a particular enzyme or target is more difficult than inhibiting it. A study suggests that a lector antibody (AL002) can bind and activate TREM2. The mouse version of the antibody, AL002a, binds to and activates TREM2. In cell culture, AL002a treatment increased the phosphorylation of Syk, a downstream effector of TREM2 signaling. Another study has also reported that a similar monoclonal antibody developed in Germany is said to activate TREM2 signaling by boosting the phospho-Syk signaling. Anti-TREM2 antibodies could perhaps serve as biomarkers, as well as therapeutic agents (Wang et al., 2020).
3. Recent developments
June 7, 2021, can be termed a historic day in the therapeutic sector of AD as aducanumab, a monoclonal antibody was approved by the Food and Drug Administration (FDA) as a drug of choice to treat AD. The drug is branded as Aduhelm and is licensed by Biogen and Eisai pharmaceutical companies under collaborative development. The FDA has approved this therapy under the accelerated approval pathway to benefit patients with serious neurodegenerative diseases following the priority review. Aduhelm works by targeting the Aβ in the brain and reduces the amount of Aβ plaques, potentially slowing down the neurodegeneration and progression of the disease (Schneider, 2020; Sevigny et al., 2016).
4. Conclusion
Several studies support the amyloidal theory that Aβ is the starter to a complex number of pathological events in the brain concerning AD. Aβ starts to emerge long before the clinical symptoms appear physically. The deposition of Aβ not only causes hindrance to the signaling system but also triggers cerebral deficits, calcium homeostasis, and cognition problems. Unfortunately, the research on Aβ plaques as a target for AD has been on a downward graph because of the failure of drugs in clinical trials. Even so, the re-launch of aducanumab as an anti-amyloid drug has stirred up this theory once again. The difficulty in pacing up with molecular knowledge is still evident as we still depend heavily on classical pathology. However, studies are done at a faster pace to identify the exact pathology behind AD suggesting that it is feasible to provide new targets for drug discovery and development directed toward the Aβ accumulation. In this article, we listed down the targets that play various roles in plaque formation, which could help design effective drugs for AD.
The best possible way to eliminate Aβ pathology is to prevent it from accumulating in the brain. The outcome that occurs on removing these pathological lesions in AD patients should be intensely focussed in the research studies.
Ethical Considerations
Compliance with ethical guidelines
There were no ethical considerations to be considered in this research.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Authors' contributions
All authors equally contributed to preparing this article.
Conflict of interest
The authors declared no conflict of interest
References
Adalbert, R., Gilley, J., & Coleman, M. P. (2007). Abeta, tau and ApoE4 in Alzheimer's disease: The axonal connection. Trends in Molecular Medicine, 13(4), 135–142. [DOI:10.1016/j.molmed.2007.02.004] [PMID]
Adler, P., Mayne, J., Walker, K., Ning, Z., & Figeys, D. (2019). Therapeutic targeting of casein kinase 1δ/ε in an alzheimer's disease mouse model. Journal of Proteome Research, 18(9), 3383–3393. [DOI:10.1021/acs.jproteome.9b00312] [PMID]
Ambrée, O., Richter, H., Sachser, N., Lewejohann, L., Dere, E., & de Souza Silva, M. A., et al. (2009). Levodopa ameliorates learning and memory deficits in a murine model of Alzheimer's disease. Neurobiology of Aging, 30(8), 1192–1204. [DOI:10.1016/j.neurobiolaging.2007.11.010] [PMID]
Ashraf, G. M., Chibber, S., Mohammad, Zaidi, S. K., Tabrez, S., & Ahmad, A., et al. (2016). Recent updates on the association between alzheimer's disease and vascular dementia. Medicinal Chemistry, 12(3), 226–237. [DOI:10.2174/1573406411666151030111820] [PMID]
Bano, S., Rasheed, M. A., Jamil, F., Ibrahim, M., & Kanwal, S. (2019). In silico identification of novel apolipoprotein e4 inhibitor for alzheimer's disease therapy. Current Computer-Aided Drug Design, 15(1), 97–103. [DOI:10.2174/1573409914666181008164209] [PMID]
Baranger, K., Marchalant, Y., Bonnet, A. E., Crouzin, N., Carrete, A., & Paumier, J. M., et al. (2016). MT5-MMP is a new pro-amyloidogenic proteinase that promotes amyloid pathology and cognitive decline in a transgenic mouse model of Alzheimer's disease. Cellular and Molecular Life Sciences, 73(1), 217–236. [DOI:10.1007/s00018-015-1992-1] [PMID] [PMCID]
Bartus, R. T., Baker, K. L., Heiser, A. D., Sawyer, S. D., Dean, R. L., & Elliott, P. J., et al. (1994). Postischemic administration of AK275, a calpain inhibitor, provides substantial protection against focal ischemic brain damage. Journal of Cerebral Blood Flow and Metabolism, 14(4), 537–544. [DOI:10.1038/jcbfm.1994.67] [PMID]
Basi, G. S., Hemphill, S., Brigham, E. F., Liao, A., Aubele, D. L., & Baker, J., et al. (2010). Amyloid precursor protein selective gamma-secretase inhibitors for treatment of Alzheimer's disease. Alzheimer's Research & Therapy, 2(6), 36. [DOI:10.1186/alzrt60] [PMID] [PMCID]
Bavishi, C., Messerli, F. H., Kadosh, B., Ruilope, L. M., & Kario, K. (2015). Role of neprilysin inhibitor combinations in hypertension: Insights from hypertension and heart failure trials. European Heart Journal, 36(30), 1967–1973. [DOI:10.1093/eurheartj/ehv142] [PMID]
Benn, C. L., & Dawson, L. A. (2020). Clinically precedented protein kinases: Rationale for their use in neurodegenerative disease. Frontiers in Aging Neuroscience, 12, 242.[DOI:10.3389/fnagi.2020.00242] [PMID] [PMCID]
Benny, A., & Thomas, J. (2019). Essential oils as treatment strategy for alzheimer's disease: Current and future perspectives. Planta Medica, 85(3), 239–248. [DOI:10.1055/a-0758-0188] [PMID]
Blain, J. F., Bursavich, M. G., Freeman, E. A., Hrdlicka, L. A., Hodgdon, H. E., & Chen, T., et al. (2016). Characterization of FRM-36143 as a new γ-secretase modulator for the potential treatment of familial Alzheimer's disease. Alzheimer's Research & Therapy, 8(1), 34. [DOI:10.1186/s13195-016-0199-5] [PMID] [PMCID]
Blume, T., Filser, S., Jaworska, A., Blain, J. F., Koenig, G., & Moschke, K., et al. (2018). BACE1 inhibitor MK-8931 alters formation but not stability of dendritic spines. Frontiers in Aging Neuroscience, 10, 229. [DOI:10.3389/fnagi.2018.00229] [PMID] [PMCID]
Boehm-Cagan, A., & Michaelson, D. M. (2014). Reversal of apoE4-driven brain pathology and behavioral deficits by bexarotene. The Journal of Neuroscience, 34(21), 7293–7301. [DOI:10.1523/JNEUROSCI.5198-13.2014] [PMID] [PMCID]
Branca, C., Shaw, D. M., Belfiore, R., Gokhale, V., Shaw, A. Y., & Foley, C., et al. (2017). Dyrk1 inhibition improves Alzheimer's disease-like pathology. Aging Cell, 16(5), 1146–1154. [DOI:10.1111/acel.12648] [PMID] [PMCID]
Brkic, M., Balusu, S., Libert, C., & Vandenbroucke, R. E. (2015). Friends or foes: Matrix metalloproteinases and their multifaceted roles in neurodegenerative diseases. Mediators of Inflammation, 2015, 620581. [DOI:10.1155/2015/620581] [PMID] [PMCID]
Burgy, G., Tahtouh, T., Durieu, E., Foll-Josselin, B., Limanton, E., & Meijer, L., et al. (2013). Chemical synthesis and biological validation of immobilized protein kinase inhibitory Leucettines. European Journal of Medicinal Chemistry, 62, 728–737. [DOI:10.1016/j.ejmech.2013.01.035] [PMID]
Carmona, S., Zahs, K., Wu, E., Dakin, K., Bras, J., & Guerreiro, R. (2018). The role of TREM2 in alzheimer's disease and other neurodegenerative disorders. The Lancet. Neurology, 17(8), 721–730. [DOI:10.1016/S1474-4422(18)30232-1] [PMID]
Cebers, G., Alexander, R. C., Haeberlein, S. B., Han, D., Goldwater, R., & Ereshefsky, L., et al. (2017). AZD3293: Pharmacokinetic and pharmacodynamic effects in healthy subjects and patients with alzheimer's disease. Journal of Alzheimer's Disease, 55(3), 1039–1053. [DOI:10.3233/JAD-160701] [PMID]
Chaudhary, A., Maurya, P. K., Yadav, B. S., Singh, S., & Mani, A. (2018). Current therapeutic targets for alzheimer’s disease. Journal of Biomedicine, 3, 74-84. [DOI:10.7150/jbm.26783]
Chauhan, A., Chauhan, V. P., Murakami, N., Brockerhoff, H., & Wisniewski, H. M. (1993). Amyloid beta-protein stimulates casein kinase I and casein kinase II activities. Brain Research, 629(1), 47–52. [DOI:10.1016/0006-8993(93)90479-7] [PMID]
Cheng, J., Guo, X., Zhang, T., Zhong, L., Bu, G., & Chen, X. (2016). TREMs in alzheimer's disease: Genetic and clinical investigations. Clinica Chimica Acta; International Journal of Clinical Chemistry, 463, 88–95. [DOI:10.1016/j.cca.2016.10.022] [PMID]
Cho, D. H., Lee, E. J., Kwon, K. J., Shin, C. Y., Song, K. H., & Park, J. H., et al. (2013). Troglitazone, a thiazolidinedione, decreases tau phosphorylation through the inhibition of cyclin-dependent kinase 5 activity in SH-SY5Y neuroblastoma cells and primary neurons. Journal of Neurochemistry, 126(5), 685–695. [DOI:10.1111/jnc.12264] [PMID]
Chon, H. J., Bae, K. J., Lee, Y., & Kim, J. (2015). The casein kinase 2 inhibitor, CX-4945, as an anti-cancer drug in treatment of human hematological malignancies. Frontiers in Pharmacology, 6, 70. [DOI:10.3389/fphar.2015.00070] [PMID] [PMCID]
Cicenas, J., Kalyan, K., Sorokinas, A., Stankunas, E., Levy, J., & Meskinyte, I., et al. (2015). Roscovitine in cancer and other diseases. Annals of Translational Medicine, 3(10), 135. [DOI:10.3978/j.issn.2305-5839.2015.03.61]
Coimbra, J. R. M., Marques, D. F. F., Baptista, S. J., Pereira, C. M. F., Moreira, P. I., & Dinis, T. C. P., et al. (2018). Highlights in BACE1 inhibitors for alzheimer's disease treatment. Frontiers in Chemistry, 6, 178. [DOI:10.3389/fchem.2018.00178] [PMID] [PMCID]
Cole, S. L., & Vassar, R. (2007). The alzheimer's disease beta-secretase enzyme, BACE1. Molecular Neurodegeneration, 2, 22. [DOI:10.1186/1750-1326-2-22] [PMID] [PMCID]
Coll, R. C., Robertson, A. A., Chae, J. J., Higgins, S. C., Muñoz-Planillo, R., & Inserra, M. C., et al. (2015). A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nature Medicine, 21(3), 248–255. [DOI:10.1038/nm.3806] [PMID] [PMCID]
Coman, H., & Nemeş, B. (2017). New therapeutic targets in alzheimer’s disease. International Journal of Gerontology, 11(1), 2-6. [DOI:10.1016/j.ijge.2016.07.003]
Cross, A. J., Crow, T. J., Perry, E. K., Perry, R. H., Blessed, G., & Tomlinson, B. E. (1981). Reduced dopamine-beta-hydroxylase activity in Alzheimer's disease. British Medical Journal, 282(6258), 93–94. [DOI:10.1136/bmj.282.6258.93] [PMID] [PMCID]
Cruz, J. C., & Tsai, L. H. (2004). Cdk5 deregulation in the pathogenesis of Alzheimer's disease. Trends in Molecular Medicine, 10(9), 452–458. [DOI:10.1016/j.molmed.2004.07.001] [PMID]
Das, B., & Yan, R. (2017). Role of BACE1 in alzheimer's synaptic function. Translational Neurodegeneration, 6, 23. [DOI:10.1186/s40035-017-0093-5] [PMID] [PMCID]
De Strooper B. (2007). Loss-of-function presenilin mutations in Alzheimer disease. Talking point on the role of presenilin mutations in Alzheimer disease. EMBO Reports, 8(2), 141–146.[DOI:10.1038/sj.embor.7400897] [PMID] [PMCID]
De Strooper, B., Iwatsubo, T., & Wolfe, M. S. (2012). Presenilins and γ-secretase: Structure, function, and role in alzheimer disease. Cold Spring Harbor Perspectives in Medicine, 2(1), a006304. [DOI:10.1101/cshperspect.a006304] [PMID] [PMCID]
Dhavan, R., & Tsai, L. H. (2001). A decade of CDK5. Nature reviews. Molecular Cell Biology, 2(10), 749–759.[DOI:10.1038/35096019] [PMID]
Du, X., Wang, X., & Geng, M. (2018). Alzheimer's disease hypothesis and related therapies. Translational Neurodegeneration, 7, 2. [DOI:10.1186/s40035-018-0107-y] [PMID] [PMCID]
Duits, F. H., Hernandez-Guillamon, M., Montaner, J., Goos, J. D., Montañola, A., & Wattjes, M. P., et al. (2015). Matrix metalloproteinases in alzheimer's disease and concurrent cerebral microbleeds. Journal of Alzheimer's Disease, 48(3), 711–720. [DOI:10.3233/JAD-143186] [PMID]
Eckman, E. A., & Eckman, C. B. (2005). Abeta-degrading enzymes: Modulators of alzheimer's disease pathogenesis and targets for therapeutic intervention. Biochemical Society Transactions, 33(Pt 5), 1101–1105. [DOI:10.1042/BST20051101] [PMID]
Edwards F. A. (2019). A unifying hypothesis for alzheimer's disease: From plaques to neurodegeneration. Trends in Neurosciences, 42(5), 310–322. [DOI:10.1016/j.tins.2019.03.003] [PMID]
Egan, M. F., Kost, J., Tariot, P. N., Aisen, P. S., Cummings, J. L., & Vellas, B.,et al. (2018). Randomized trial of verubecestat for mild-to-moderate alzheimer's disease. The New England Journal of Medicine, 378(18), 1691–1703. [DOI:10.1056/NEJMoa1706441] [PMID] [PMCID]
Eketjäll, S., Janson, J., Kaspersson, K., Bogstedt, A., Jeppsson, F., & Fälting, J., et al. (2016). AZD3293: A novel, orally active bace1 inhibitor with high potency and permeability and markedly slow off-rate kinetics. Journal of Alzheimer's Disease, 50(4), 1109–1123. [DOI:10.3233/JAD-150834] [PMID] [PMCID]
El-Amouri, S. S., Zhu, H., Yu, J., Marr, R., Verma, I. M., & Kindy, M. S. (2008). Neprilysin: An enzyme candidate to slow the progression of Alzheimer's disease. The American Journal of Pathology, 172(5), 1342–1354. [DOI:10.2353/ajpath.2008.070620] [PMID] [PMCID]
Evin, G., & Hince, C. (2013). BACE1 as a therapeutic target in Alzheimer's disease: Rationale and current status. Drugs & Aging, 30(10), 755–764. [DOI:10.1007/s40266-013-0099-3] [PMID]
Feki, A., & Hibaoui, Y. (2018). DYRK1A protein, a promising therapeutic target to improve cognitive deficits in down syndrome. Brain Sciences, 8(10), 187. [DOI:10.3390/brainsci8100187] [PMID] [PMCID]
Feng, Y. S., Tan, Z. X., Wu, L. Y., Dong, F., & Zhang, F. (2020). The involvement of NLRP3 inflammasome in the treatment of Alzheimer's disease. Ageing Research Reviews, 64, 101192.[DOI:10.1016/j.arr.2020.101192] [PMID]
Ferrer, I., Barrachina, M., Puig, B., Martínez de Lagrán, M., Martí, E., & Avila, J., et al. (2005). Constitutive Dyrk1A is abnormally expressed in Alzheimer disease, Down syndrome, Pick disease, and related transgenic models. Neurobiology of Disease, 20(2), 392–400. [DOI:10.1016/j.nbd.2005.03.020] [PMID]
Fischer, A., Sananbenesi, F., Schrick, C., Spiess, J., & Radulovic, J. (2002). Cyclin-dependent kinase 5 is required for associative learning. The Journal of Neuroscience, 22(9), 3700–3707. [DOI:10.1523/JNEUROSCI.22-09-03700.2002] [PMID] [PMCID]
Flajolet, M., He, G., Heiman, M., Lin, A., Nairn, A. C., & Greengard, P. (2007). Regulation of alzheimer's disease amyloid-beta formation by casein kinase I. Proceedings of the National Academy of Sciences of the United States of America, 104(10), 4159–4164. [DOI:10.1073/pnas.0611236104] [PMID] [PMCID]
Fragkouli, A., Tsilibary, E. C., & Tzinia, A. K. (2014). Neuroprotective role of MMP-9 overexpression in the brain of alzheimer's 5xFAD mice. Neurobiology of Disease, 70, 179–189.[DOI:10.1016/j.nbd.2014.06.021] [PMID]
Galceran, J., de Graaf, K., Tejedor, F. J., & Becker, W. (2003). The MNB/DYRK1A protein kinase: Genetic and biochemical properties. Journal of Neural Transmission. Supplementum, (67), 139–148. [DOI:10.1007/978-3-7091-6721-2_12] [PMID]
Gall, F. M., Hohl, D., Frasson, D., Wermelinger, T., Mittl, P. R. E., & Sievers, M., et al. (2019). Drug design inspired by nature: Crystallographic detection of an auto-tailored protease inhibitor template. Angewandte Chemie, 58(12), 4051–4055. [DOI:10.1002/anie.201812348] [PMID]
Gopalakrishnan, A., Shankarappa, S. A., & Rajanikant, G. K. (2019). Hydrogel scaffolds: Towards restitution of ischemic stroke-injured brain. Translational Stroke Research, 10(1), 1–18. [DOI:10.1007/s12975-018-0655-6] [PMID]
Gratuze, M., Leyns, C. E. G., & Holtzman, D. M. (2018). New insights into the role of TREM2 in alzheimer's disease. Molecular Neurodegeneration, 13(1), 66. [DOI:10.1186/s13024-018-0298-9] [PMID] [PMCID]
Halle, A., Hornung, V., Petzold, G. C., Stewart, C. R., Monks, B. G., & Reinheckel, T., et al. (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nature Immunology, 9(8), 857–865. [DOI:10.1038/ni.1636] [PMID] [PMCID]
Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., & Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature, 493(7434), 674–678. [DOI:10.1038/nature11729] [PMID] [PMCID]
Howard, R., Zubko, O., Bradley, R., Harper, E., Pank, L., & O'Brien, J., et al. (2020). Minocycline at 2 different dosages vs placebo for patients with mild alzheimer disease: A randomized clinical trial. JAMA Neurology, 77(2), 164–174. [DOI:10.1001/jamaneurol.2019.3762] [PMID] [PMCID]
Hu, X., Das, B., Hou, H., He, W., & Yan, R. (2018). BACE1 deletion in the adult mouse reverses preformed amyloid deposition and improves cognitive functions. The Journal of Experimental Medicine, 215(3), 927–940. [DOI:10.1084/jem.20171831] [PMID] [PMCID]
Huang, S. M., Mouri, A., Kokubo, H., Nakajima, R., Suemoto, T., & Higuchi, M.,et al. (2006). Neprilysin-sensitive synapse-associated amyloid-beta peptide oligomers impair neuronal plasticity and cognitive function. The Journal of Biological Chemistry, 281(26), 17941–17951. [DOI:10.1074/jbc.M601372200] [PMID]
Huang, Y., Jiang, H., Chen, Y., Wang, X., Yang, Y., & Tao, J., et al. (2018). Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Molecular Medicine, 10(4), e8689. [DOI:10.15252/emmm.201708689] [PMID] [PMCID]
Huber, R. J., & O'Day, D. H. (2012). The cyclin-dependent kinase inhibitor roscovitine inhibits kinase activity, cell proliferation, multicellular development, and Cdk5 nuclear translocation in Dictyostelium discoideum. Journal of Cellular Biochemistry, 113(3), 868–876. [DOI:10.1002/jcb.23417] [PMID]
Imbimbo, B. P., & Watling, M. (2019). Investigational BACE inhibitors for the treatment of Alzheimer's disease. Expert Opinion On Investigational Drugs, 28(11), 967–975. [DOI:10.1080/13543784.2019.1683160] [PMID]
Jay, T. R., von Saucken, V. E., & Landreth, G. E. (2017). TREM2 in neurodegenerative diseases. Molecular Neurodegeneration, 12(1), 56. [DOI:10.1186/s13024-017-0197-5] [PMID] [PMCID]
Jiang, H., He, H., Chen, Y., Huang, W., Cheng, J., & Ye, J., et al. (2017). Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. The Journal of Experimental Medicine, 214(11), 3219–3238. [DOI:10.1084/jem.20171419] [PMID] [PMCID]
Karanfilian, L., Tosto, M. G., & Malki, K. (2020). The role of TREM2 in alzheimer's disease; evidence from transgenic mouse models. Neurobiology of Aging, 86, 39–53.[DOI:10.1016/j.neurobiolaging.2019.09.004] [PMID]
Kemppainen, N., Laine, M., Laakso, M. P., Kaasinen, V., Någren, K., & Vahlberg, T., et al. (2003). Hippocampal dopamine D2 receptors correlate with memory functions in Alzheimer's disease. The European Journal of Neuroscience, 18(1), 149–154. [DOI:10.1046/j.1460-9568.2003.02716.x] [PMID]
Kennedy, M. E., Stamford, A. W., Chen, X., Cox, K., Cumming, J. N., & Dockendorf, M. F., et al. (2016). The BACE1 inhibitor verubecestat (MK-8931) reduces CNS β-amyloid in animal models and in alzheimer's disease patients. Science Translational Medicine, 8(363), 363ra150. [DOI:10.1126/scitranslmed.aad9704] [PMID]
Khalil, H. S., Mitev, V., Vlaykova, T., Cavicchi, L., & Zhelev, N. (2015). Discovery and development of Seliciclib. How systems biology approaches can lead to better drug performance. Journal of Biotechnology, 202, 40–49. [DOI:10.1016/j.jbiotec.2015.02.032] [PMID]
Kounnas, M. Z., Danks, A. M., Cheng, S., Tyree, C., Ackerman, E., & Zhang, X., et al. (2010). Modulation of gamma-secretase reduces beta-amyloid deposition in a transgenic mouse model of alzheimer's disease. Neuron, 67(5), 769–780. [DOI:10.1016/j.neuron.2010.08.018] [PMID] [PMCID]
Kuo, L. M., Kuo, C. Y., Lin, C. Y., Hung, M. F., Shen, J. J., & Hwang, T. L. (2014). Intracellular glutathione depletion by oridonin leads to apoptosis in hepatic stellate cells. Molecules, 19(3), 3327–3344. [DOI:10.3390/molecules19033327] [PMID] [PMCID]
Lahiri, D. K., Maloney, B., Long, J. M., & Greig, N. H. (2014). Lessons from a BACE1 inhibitor trial: Off-site but not off base. Alzheimer's & Dementia, 10(5 Suppl), S411–S419. [DOI:10.1016/j.jalz.2013.11.004] [PMID] [PMCID]
Laird, F. M., Cai, H., Savonenko, A. V., Farah, M. H., He, K., & Melnikova, T., et al. (2005). BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. The Journal of Neuroscience, 25(50), 11693–11709. [DOI:10.1523/JNEUROSCI.2766-05.2005] [PMID] [PMCID]
Lamkanfi, M., & Dixit, V. M. (2017). A new lead to NLRP3 inhibition. The Journal of Experimental Medicine, 214(11), 3147–3149. [DOI:10.1084/jem.20171848] [PMID] [PMCID]
Lee, H. J., Woo, H., Lee, H. E., Jeon, H., Ryu, K. Y., & Nam, J. H., et al. (2020). The novel DYRK1A inhibitor KVN93 regulates cognitive function, amyloid-beta pathology, and neuroinflammation. Free Radical Biology & Medicine, 160, 575–595. [DOI:10.1016/j.freeradbiomed.2020.08.030] [PMID]
Liu, C. C., Liu, C. C., Kanekiyo, T., Xu, H., & Bu, G. (2013). Apolipoprotein E and alzheimer disease: Risk, mechanisms and therapy. Nature Reviews. Neurology, 9(2), 106–118.[DOI:10.1038/nrneurol.2012.263] [PMID] [PMCID]
Liu, C. C., Zhao, N., Fu, Y., Wang, N., Linares, C., & Tsai, C. W., et al. (2017). ApoE4 accelerates early seeding of amyloid pathology. Neuron, 96(5), 1024–1032.e3. [DOI:10.1016/j.neuron.2017.11.013] [PMID] [PMCID]
Liu, F., Su, Y., Li, B., Zhou, Y., Ryder, J., & Gonzalez-DeWhitt, P., et al. (2003). Regulation of amyloid precursor protein (APP) phosphorylation and processing by p35/Cdk5 and p25/Cdk5. FEBS Letters, 547(1-3), 193–196. [DOI:10.1016/s0014-5793(03)00714-2] [PMID]
Liu, M., Kou, L., Bin, Y., Wan, L., & Xiang, J. (2016). Complicated function of dopamine in Aβ-related neurotoxicity: Dual interactions with Tyr10 and SNK(26-28) of Aβ. Journal of Inorganic Biochemistry, 164, 119–128. [DOI:10.1016/j.jinorgbio.2016.09.007] [PMID]
Lorenzl, S., Albers, D. S., Relkin, N., Ngyuen, T., Hilgenberg, S. L., & Chirichigno, J., et al. (2003). Increased plasma levels of matrix metalloproteinase-9 in patients with alzheimer's disease. Neurochemistry International, 43(3), 191–196. [DOI:10.1016/s0197-0186(03)00004-4] [PMID]
Madani, R., Poirier, R., Wolfer, D. P., Welzl, H., Groscurth, P., & Lipp, H. P., et al. (2006). Lack of neprilysin suffices to generate murine amyloid-like deposits in the brain and behavioral deficit in vivo. Journal of Neuroscience Research, 84(8), 1871–1878. [DOI:10.1002/jnr.21074] [PMID]
Madav, Y., Wairkar, S., & Prabhakar, B. (2019). Recent therapeutic strategies targeting beta amyloid and tauopathies in Alzheimer's disease. Brain Research Bulletin, 146, 171–184.[DOI:10.1016/j.brainresbull.2019.01.004] [PMID]
Manakadan, A. A., Suja, S. T., Silvipriya, K. S., & Chithra, J. (2015). A Computational in silico approach for identification of indian herbs for the treatment of alzheimer’s disease. Research Journal of Pharmaceutical, Biological and Chemical Sciences, 6(1), 414-422. [Link]
Marchetti, C., Swartzwelter, B., Koenders, M. I., Azam, T., Tengesdal, I. W., & Powers, N., et al. (2018). NLRP3 inflammasome inhibitor OLT1177 suppresses joint inflammation in murine models of acute arthritis. Arthritis Research & Therapy, 20(1), 169.[DOI:10.1186/s13075-018-1664-2] [PMID] [PMCID]
Marr, R. A., & Hafez, D. M. (2014). Amyloid-beta and alzheimer’s disease: The role of neprilysin-2 in amyloid-beta clearance. Frontiers in Aging Neuroscience, 6, 187. [DOI:10.3389/fnagi.2014.00187]
Martone, R. L., Zhou, H., Atchison, K., Comery, T., Xu, J. Z., & Huang, X., et al. (2009). Begacestat (GSI-953): A novel, selective thiophene sulfonamide inhibitor of amyloid precursor protein gamma-secretase for the treatment of Alzheimer's disease. The Journal of Pharmacology and Experimental Therapeutics, 331(2), 598–608. [DOI:10.1124/jpet.109.152975] [PMID]
Martorana, A., & Koch, G. (2014). Is dopamine involved in Alzheimer’s disease? Frontiers in Aging Neuroscience, 6, 252. [DOI:10.3389/fnagi.2014.00252]
Maruyama, M., Higuchi, M., Takaki, Y., Matsuba, Y., Tanji, H., & Nemoto, M., et al. (2005). Cerebrospinal fluid neprilysin is reduced in prodromal Alzheimer's disease. Annals of Neurology, 57(6), 832–842.[DOI:10.1002/ana.20494] [PMID]
Melchior, B., Mittapalli, G. K., Lai, C., Duong-Polk, K., Stewart, J., & Güner, B., et al. (2019). Tau pathology reduction with SM07883, a novel, potent, and selective oral DYRK1A inhibitor: A potential therapeutic for alzheimer's disease. Aging Cell, 18(5), e13000.[DOI:10.1111/acel.13000] [PMID] [PMCID]
Menn, B., Bach, S., Blevins, T. L., Campbell, M., Meijer, L., & Timsit, S. (2010). Delayed treatment with systemic (S)-roscovitine provides neuroprotection and inhibits in vivo CDK5 activity increase in animal stroke models. Plos One, 5(8), e12117.[DOI:10.1371/journal.pone.0012117] [PMID] [PMCID]
Morris, J. C., Roe, C. M., Xiong, C., Fagan, A. M., Goate, A. M., & Holtzman, D. M., et al. (2010). APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Annals of Neurology, 67(1), 122–131.[DOI:10.1002/ana.21843] [PMID] [PMCID]
Namba, Y., & Ikeda, K. (1991). [Apolipoprotein B immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in senile dementia of alzheimer type (Japanese)]. Rinsho shinkeigaku = Clinical neurology, 31(8), 826–830. [PMID]
Neumann, U., Ufer, M., Jacobson, L. H., Rouzade-Dominguez, M. L., Huledal, G., & Kolly, C., et al. (2018). The BACE1 inhibitor CNP520 for prevention trials in alzheimer's disease. EMBO Molecular Medicine, 10(11), e9316. [DOI:10.15252/emmm.201809316] [PMID] [PMCID]
Nobili, A., Latagliata, E. C., Viscomi, M. T., Cavallucci, V., Cutuli, D., & Giacovazzo, G., et al. (2017). Dopamine neuronal loss contributes to memory and reward dysfunction in a model of alzheimer's disease. Nature Communications, 8, 14727. [DOI:10.1038/ncomms14727] [PMID] [PMCID]
Octave J. N. (1995). The amyloid peptide and its precursor in Alzheimer's disease. Reviews in the Neurosciences, 6(4), 287–316. [DOI:10.1515/REVNEURO.1995.6.4.287] [PMID]
Pan, X., Kaminga, A. C., Wen, S. W., Wu, X., Acheampong, K., & Liu, A. (2019). Dopamine and dopamine receptors in alzheimer's disease: A systematic review and network meta-analysis. Frontiers in Aging Neuroscience, 11, 175. [DOI:10.3389/fnagi.2019.00175] [PMID] [PMCID]
Panza, F., Frisardi, V., Imbimbo, B. P., Capurso, C., Logroscino, G., & Sancarlo, D.,et al. (2010). Review: γ-secretase inhibitors for the treatment of alzheimer's disease: The current state. Cns Neuroscience & Therapeutics, 16(5), 272–284. [DOI:10.1111/j.1755-5949.2010.00164.x] [PMID] [PMCID]
Pathak, A., Rohilla, A., Gupta, T., Akhtar, M. J., Haider, M. R., & Sharma, K., et al. (2018). DYRK1A kinase inhibition with emphasis on neurodegeneration: A comprehensive evolution story-cum-perspective. European Journal of Medicinal Chemistry, 158, 559–592. [DOI:10.1016/j.ejmech.2018.08.093] [PMID]
Pavo, N., Prausmüller, S., Bartko, P. E., Goliasch, G., & Hülsmann, M. (2020). Neprilysin as a biomarker: Challenges and opportunities. Cardiac Failure Review, 6, e23. [DOI:10.15420/cfr.2019.21] [PMID] [PMCID]
Rajendran, L., Schneider, A., Schlechtingen, G., Weidlich, S., Ries, J., Braxmeier, T., & Schwille, P., et al. (2008). Efficient inhibition of the alzheimer's disease beta-secretase by membrane targeting. Science, 320(5875), 520–523. [DOI:10.1126/science.1156609] [PMID]
Reeves, S., McLachlan, E., Bertrand, J., D'Antonio, F., Brownings, S., & Nair, A., et al. (2017). Therapeutic window of dopamine D2/3 receptor occupancy to treat psychosis in alzheimer's disease. Brain, 140(4), 1117–1127. [DOI:10.1093/brain/aww359] [PMID]
Reinhardt, L., Kordes, S., Reinhardt, P., Glatza, M., Baumann, M., & Drexler, H. C. A., et al. (2019). Dual inhibition of GSK3β and CDK5 protects the cytoskeleton of neurons from neuroinflammatory-mediated degeneration in vitro and in vivo. Stem Cell Reports, 12(3), 502–517. [DOI:10.1016/j.stemcr.2019.01.015] [PMID] [PMCID]
Rinne, J. O., Säkö, E., Paljärvi, L., Mölsä, P. K., & Rinne, U. K. (1986). Brain dopamine D-2 receptors in senile dementia. Journal of Neural Transmission, 65(1), 51–62. [DOI:10.1007/BF01249611] [PMID]
Rizos, C. V., Elisaf, M. S., Mikhailidis, D. P., & Liberopoulos, E. N. (2009). How safe is the use of thiazolidinediones in clinical practice?. Expert Opinion on Drug Safety, 8(1), 15–32.[DOI:10.1517/14740330802597821] [PMID]
Rizzi, L., Rosset, I., & Roriz-Cruz, M. (2014). Global epidemiology of dementia: Alzheimer's and vascular types. BioMed Research International, 2014, 908915. [DOI:10.1155/2014/908915] [PMID] [PMCID]
Rodrigues, R. P., & Silva, C. H. T. P. (2017). Discovery of potential neurodegenerative inhibitors in alzheimer’s disease by casein kinase 1 structure-based virtual screening. Medicinal Chemistry Research, 26(12), 3274-3285. [DOI:10.1007/s00044-017-2020-9]
Rosenberg, P. B., Nowrangi, M. A., & Lyketsos, C. G. (2015). Neuropsychiatric symptoms in Alzheimer's disease: What might be associated brain circuits?. Molecular Aspects of Medicine, 43-44, 25–37. [DOI:10.1016/j.mam.2015.05.005] [PMID] [PMCID]
Safieh, M., Korczyn, A. D., & Michaelson, D. M. (2019). ApoE4: An emerging therapeutic target for Alzheimer's disease. BMC Medicine, 17(1), 64. [DOI:10.1186/s12916-019-1299-4] [PMID] [PMCID]
Sakamoto, K., Matsuki, S., Matsuguma, K., Yoshihara, T., Uchida, N., & Azuma, F., et al. (2017). BACE1 inhibitor lanabecestat (AZD3293) in a phase 1 study of healthy japanese subjects: Pharmacokinetics and effects on plasma and cerebrospinal fluid Aβ peptides. Journal of Clinical Pharmacology, 57(11), 1460–1471. [DOI:10.1002/jcph.950] [PMID]
Schiering, N., D'Arcy, A., Villard, F., Ramage, P., Logel, C., & Cumin, F., et al. (2016). Structure of neprilysin in complex with the active metabolite of sacubitril. Scientific Reports, 6, 27909. [DOI:10.1038/srep27909] [PMID] [PMCID]
Schneider L. (2020). A resurrection of aducanumab for Alzheimer's disease. The Lancet. Neurology, 19(2), 111–112.[DOI:10.1016/S1474-4422(19)30480-6] [PMID]
Scott, J. D., Li, S. W., Brunskill, A. P., Chen, X., Cox, K., & Cumming, J. N., et al. (2016). Discovery of the 3-Imino-1,2,4-thiadiazinane 1,1-Dioxide derivative verubecestat (MK-8931)-A β-site amyloid precursor protein cleaving enzyme 1 Inhibitor for the treatment of alzheimer's disease. Journal of Medicinal Chemistry, 59(23), 10435–10450. [DOI:10.1021/acs.jmedchem.6b00307] [PMID]
Seo, J., Kritskiy, O., Watson, L. A., Barker, S. J., Dey, D., & Raja, W. K., et al. (2017). Inhibition of p25/Cdk5 attenuates tauopathy in mouse and iPSC models of frontotemporal dementia. The Journal of Neuroscience, 37(41), 9917–9924. [DOI:10.1523/JNEUROSCI.0621-17.2017] [PMID] [PMCID]
Serrano-Pozo, A., Frosch, M. P., Masliah, E., & Hyman, B. T. (2011). Neuropathological alterations in alzheimer disease. Cold Spring Harbor Perspectives in Medicine, 1(1), a006189. [DOI:10.1101/cshperspect.a006189] [PMID] [PMCID]
Sevigny, J., Chiao, P., Bussière, T., Weinreb, P. H., Williams, L., & Maier, M., et al. (2016). The antibody aducanumab reduces Aβ plaques in alzheimer's disease. Nature, 537(7618), 50–56. [DOI:10.1038/nature19323] [PMID]
Shah, K., & Lahiri, D. K. (2014). Cdk5 activity in the brain - multiple paths of regulation. Journal of Cell Science, 127(Pt 11), 2391–2400. [DOI:10.1242/jcs.147553] [PMID] [PMCID]
Shankarappa, S. A., Tsui, J. H., Kim, K. N., Reznor, G., Dohlman, J. C., & Langer, R., et al. (2012). Prolonged nerve blockade delays the onset of neuropathic pain. Proceedings of the National Academy of Sciences of the United States of America, 109(43), 17555–17560. [DOI:10.1073/pnas.1214634109] [PMID] [PMCID]
Shukla, V., Skuntz, S., & Pant, H. C. (2012). Deregulated Cdk5 activity is involved in inducing Alzheimer's disease. Archives of Medical Research, 43(8), 655–662. [DOI:10.1016/j.arcmed.2012.10.015] [PMID] [PMCID]
Song, L., Pei, L., Yao, S., Wu, Y., & Shang, Y. (2017). NLRP3 inflammasome in neurological diseases, from functions to therapies. Frontiers in Cellular Neuroscience, 11, 63. [DOI:10.3389/fncel.2017.00063]
Soto, M., Reynish, E., Nourhashémi, F., & Vellas, B. (2007). [Clinical aspects of Alzheimer disease (French)]. Presse Medicale, 36(10 Pt 2), 1491–1499. [DOI:10.1016/j.lpm.2007.04.024] [PMID]
Souchet, B., Audrain, M., Billard, J. M., Dairou, J., Fol, R., & Orefice, N. S., et al. (2019). Inhibition of DYRK1A proteolysis modifies its kinase specificity and rescues alzheimer phenotype in APP/PS1 mice. Acta Neuropathologica Communications, 7(1), 46. [DOI:10.1186/s40478-019-0678-6] [PMID] [PMCID]
Stotani, S., Giordanetto, F., & Medda, F. (2016). DYRK1A inhibition as potential treatment for alzheimer's disease. Future Medicinal Chemistry, 8(6), 681–696. [DOI:10.4155/fmc-2016-0013] [PMID]
Sundaram, S., Nagaraj, S., Mahoney, H., Portugues, A., Li, W., & Millsaps, K., et al. (2019). Inhibition of casein kinase 1δ/εimproves cognitive-affective behavior and reduces amyloid load in the APP-PS1 mouse model of alzheimer's disease. Scientific Reports, 9(1), 13743. [DOI:10.1038/s41598-019-50197-x] [PMID] [PMCID]
Tan, M. S., Yu, J. T., Jiang, T., Zhu, X. C., & Tan, L. (2013). The NLRP3 inflammasome in alzheimer's disease. Molecular Neurobiology, 48(3), 875–882. [DOI:10.1007/s12035-013-8475-x] [PMID]
Tang, W., Lv, B., Yang, B., Chen, Y., Yuan, F., & Ma, L., et al. (2019). TREM2 acts as a tumor suppressor in hepatocellular carcinoma by targeting the PI3K/Akt/β-catenin pathway. Oncogenesis, 8(2), 9. [DOI:10.1038/s41389-018-0115-x] [PMID] [PMCID]
Taniguchi, S., Fujita, Y., Hayashi, S., Kakita, A., Takahashi, H., & Murayama, S., et al. (2001). Calpain-mediated degradation of p35 to p25 in postmortem human and rat brains. FEBs Letters, 489(1), 46–50. [DOI:10.1016/s0014-5793(00)02431-5] [PMID]
Timmers, M., Streffer, J. R., Russu, A., Tominaga, Y., Shimizu, H., & Shiraishi, A., et al. (2018). Pharmacodynamics of atabecestat (JNJ-54861911), an oral BACE1 inhibitor in patients with early Alzheimer's disease: Randomized, double-blind, placebo-controlled study. Alzheimer's Research & Therapy, 10(1), 85. [DOI:10.1186/s13195-018-0415-6] [PMID] [PMCID]
Toldo, S., Mauro, A. G., Cutter, Z., Van Tassell, B. W., Mezzaroma, E., & Del Buono, M. G., et al. (2019). The NLRP3 inflammasome inhibitor, OLT1177 (Dapansutrile), reduces infarct size and preserves contractile function after ischemia reperfusion injury in the mouse. Journal of Cardiovascular Pharmacology, 73(4), 215–222. [DOI:10.1097/FJC.0000000000000658] [PMID]
Vassar, R., & Kandalepas, P. C. (2011). The β-secretase enzyme BACE1 as a therapeutic target for alzheimer's disease. Alzheimer's Research & Therapy, 3(3), 20. [DOI:10.1186/alzrt82] [PMID] [PMCID]
Velazquez, R., Meechoovet, B., Ow, A., Foley, C., Shaw, A., & Smith, B., et al. (2019). Chronic dyrk1 inhibition delays the onset of AD-like pathology in 3xTg-AD mice. Molecular Neurobiology, 56(12), 8364–8375. [DOI:10.1007/s12035-019-01684-9] [PMID]
Vodovar, N., Paquet, C., Mebazaa, A., Launay, J. M., Hugon, J., & Cohen-Solal, A. (2015). Neprilysin, cardiovascular, and Alzheimer's diseases: The therapeutic split?. European Heart Journal, 36(15), 902–905. [DOI:10.1093/eurheartj/ehv015] [PMID]
Wang, S., Mustafa, M., Yuede, C. M., Salazar, S. V., Kong, P., & Long, H., et al. (2020). Anti-human TREM2 induces microglia proliferation and reduces pathology in an alzheimer's disease model. The Journal of Experimental Medicine, 217(9), e20200785. [DOI:10.1084/jem.20200785] [PMID] [PMCID]
Wang, S., Yuan, Y. H., Chen, N. H., & Wang, H. B. (2019). The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson's disease. International Immunopharmacology, 67, 458–464. [DOI:10.1016/j.intimp.2018.12.019] [PMID]
Wang, W. Y., Tan, M. S., Yu, J. T., & Tan, L. (2015). Role of pro-inflammatory cytokines released from microglia in alzheimer's disease. Annals of Translational Medicine, 3(10), 136.[DOI:10.3978/j.issn.2305-5839.2015.03.49] [PMID] [PMCID]
Wang, X. X., Tan, M. S., Yu, J. T., & Tan, L. (2014). Matrix metalloproteinases and their multiple roles in alzheimer's disease. BioMed Research International, 2014, 908636. [DOI:10.1155/2014/908636] [PMID] [PMCID]
Wang, Y., Zhao, J., Guo, F. L., Gao, X., Xie, X., & Liu, S., et al. (2020). Metformin ameliorates synaptic defects in a mouse model of AD by inhibiting cdk5 activity. Frontiers in Cellular Neuroscience, 14, 170. [DOI:10.3389/fncel.2020.00170] [PMID] [PMCID]
Webster, C. I., Burrell, M., Olsson, L. L., Fowler, S. B., Digby, S., & Sandercock, A., et al. (2014). Engineering neprilysin activity and specificity to create a novel therapeutic for alzheimer's disease. Plos One, 9(8), e104001. [DOI:10.1371/journal.pone.0104001] [PMID] [PMCID]
Wilkaniec, A., Czapski, G. A., & Adamczyk, A. (2016). Cdk5 at crossroads of protein oligomerization in neurodegenerative diseases: Facts and hypotheses. Journal of Neurochemistry, 136(2), 222–233. [DOI:10.1111/jnc.13365] [PMID]
Wilkaniec, A., Gąssowska-Dobrowolska, M., Strawski, M., Adamczyk, A., & Czapski, G. A. (2018). Inhibition of cyclin-dependent kinase 5 affects early neuroinflammatory signalling in murine model of amyloid beta toxicity. Journal of Neuroinflammation, 15(1), 1. [DOI:10.1186/s12974-017-1027-y] [PMID] [PMCID]
Wolfe M. S. (2012). γ-Secretase inhibitors and modulators for alzheimer's disease. Journal of Neurochemistry, 120(Suppl 1), 89–98. [DOI:10.1111/j.1471-4159.2011.07501.x] [PMID] [PMCID]
Wolfe M. S. (2014). Unlocking truths of γ-secretase in Alzheimer's disease: What is the translational potential?. Future Neurology, 9(4), 419–429. [DOI:10.2217/fnl.14.35] [PMID] [PMCID]
Yamazaki, Y., Zhao, N., Caulfield, T. R., Liu, C. C., & Bu, G. (2019). Apolipoprotein E and alzheimer disease: Pathobiology and targeting strategies. Nature Reviews. Neurology, 15(9), 501–518. [DOI:10.1038/s41582-019-0228-7] [PMID] [PMCID]
Yan, R., Bienkowski, M. J., Shuck, M. E., Miao, H., Tory, M. C., & Pauley, A. M., et al. (1999). Membrane-anchored aspartyl protease with alzheimer's disease beta-secretase activity. Nature, 402(6761), 533–537 [DOI:10.1038/990107] [PMID]
Yang, E. J., Ahn, Y. S., & Chung, K. C. (2001). Protein kinase Dyrk1 activates cAMP response element-binding protein during neuronal differentiation in hippocampal progenitor cells. The Journal of Biological Chemistry, 276(43), 39819–39824. [DOI:10.1074/jbc.M104091200] [PMID]
Yang, S. J., Shao, G. F., Chen, J. L., & Gong, J. (2018). The NLRP3 inflammasome: An important driver of neuroinflammation in hemorrhagic stroke. Cellular and Molecular Neurobiology, 38(3), 595–603. [DOI:10.1007/s10571-017-0526-9] [PMID]
Yang, Y., Wang, H., Kouadir, M., Song, H., & Shi, F. (2019). Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death & Disease, 10(2), 128.[DOI:10.1038/s41419-019-1413-8] [PMID] [PMCID]
Yasojima, K., Kuret, J., DeMaggio, A. J., McGeer, E., & McGeer, P. L. (2000). Casein kinase 1 delta mRNA is upregulated in alzheimer disease brain. Brain Research, 865(1), 116–120. [DOI:10.1016/s0006-8993(00)02200-9] [PMID]
Yildiz-Unal, A., Korulu, S., & Karabay, A. (2015). Neuroprotective strategies against calpain-mediated neurodegeneration. Neuropsychiatric Disease and Treatment, 11, 297–310. [DOI:10.2147/NDT.S78226] [PMID] [PMCID]
Yin, J., Zhao, F., Chojnacki, J. E., Fulp, J., Klein, W. L., & Zhang, S., et al. (2018). NLRP3 inflammasome inhibitor ameliorates amyloid pathology in a mouse model of alzheimer's disease. Molecular Neurobiology, 55(3), 1977–1987.[DOI:10.1007/s12035-017-0467-9] [PMID] [PMCID]
Yin, K. J., Cirrito, J. R., Yan, P., Hu, X., Xiao, Q., & Pan, X., et al. (2006). Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. The Journal of Neuroscience, 26(43), 10939–10948. [DOI:10.1523/JNEUROSCI.2085-06.2006] [PMID] [PMCID]
Yong, V. W., Agrawal, S. M., & Stirling, D. P. (2007). Targeting MMPs in acute and chronic neurological conditions. Neurotherapeutic, 4(4), 580–589. [DOI:10.1016/j.nurt.2007.07.005] [PMID] [PMCID]
Yoshiyama, Y., Asahina, M., & Hattori, T. (2000). Selective distribution of matrix metalloproteinase-3 (MMP-3) in alzheimer's disease brain. Acta Neuropathologica, 99(2), 91–95. [DOI:10.1007/pl00007428] [PMID]
Zahid, A., Li, B., Kombe, A. J. K., Jin, T., & Tao, J. (2019). Pharmacological inhibitors of the NLRP3 inflammasome. Frontiers in Immunology, 10, 2538. [DOI:10.3389/fimmu.2019.02538] [PMID] [PMCID]
Zhang, Y. W., Thompson, R., Zhang, H., & Xu, H. (2011). APP processing in alzheimer's disease. Molecular Brain, 4, 3. [DOI:10.1186/1756-6606-4-3] [PMID] [PMCID]
Zheng, H., Cheng, B., Li, Y., Li, X., Chen, X., & Zhang, Y. W. (2018). TREM2 in alzheimer's disease: Microglial survival and energy metabolism. Frontiers in Aging Neuroscience, 10, 395. [DOI:10.3389/fnagi.2018.00395] [PMID] [PMCID]
Zwan, M. D., Bouwman, F. H., Konijnenberg, E., van der Flier, W. M., Lammertsma, A. A., & Verhey, F. R., et al. (2017). Diagnostic impact of [18F]flutemetamol PET in early-onset dementia. Alzheimer's Research & Therapy, 9(1), 2. [DOI:10.1186/s13195-016-0228-4] [PMID] [PMCID]
Alzheimer's disease (AD) is a progressive neuronal disorder that develops with age and leads to cognitive decline and thinking skills. AD has been a global concern for years, comprising 60-80% of dementia cases (Ashraf et al., 2016). The Alzheimer's association estimated that 10 million around the globe are affected by AD and the number is on the rise. India is ranked third highest in the caseload related to dementia and particularly in AD after China and the US. According to statistics, the population of the elderly in India will reach around 300 million, accounting for almost 20% of the total population of the country. The expected rise of AD patients in the world by 2050 would be from 1.6 million cases in 2015 to 4.6 million cases by 2050 (Edwards, 2019; Madav et al., 2019; Rizzi et al., 2014; Soto et al., 2007). Pathologically, AD is characterized by intracellular neurofibrillary tangles and extracellular amyloid-beta (Aβ) plaques, which are first seen in the cortex and hippocampus. This induces neuronal injury resulting in neuronal death and successively damaging the cholinergic transmission leading to loss of memory and cognition along with neurotransmitter abnormalities (Benny & Thomas, 2019; Chaudhary et al., 2018; Gopalakrishnan et al., 2019; Shankarappa et al., 2012).
Over the years, advances in the field of pathogenesis have inspired researchers to identify novel therapies concentrated more on the pathological aspects of the disease. Although the exact root cause of AD remains unclear, several factors play a major role in the progression of the disease (Madav et al., 2019). The various hypotheses developed for the pathogenesis of AD are included in Figure 1.
The Aβ hypothesis remains the most studied area of AD pathology and the preferred target for drug development. The proteolytic processing of amyloid precursor protein (APP) is said to be the central dogma of this hypothesis. APP is a transmembrane protein located on chromosome 21 in humans and is expressed in the brain at high levels and is metabolized rapidly. APP is mainly synthesized in the endoplasmic reticulum and then moves to the Golgi network where it plays a role in neural growth and repair. The proteolysis of APP occurs by amyloidogenic (pathogenic) or the non-amyloidogenic (non-pathogenic) pathway. The amyloidogenic pathway releases insoluble Aβ peptides and the aggregation of these peptides leads to plaque formation (Du et al., 2018; Edwards, 2019; Serrano-Pozo et al., 2011; Zhang et al., 2011).
2. Aβ: The miracle worker
Aβ is a peptide generated throughout life, while the formation of Aβ plaques is a neuropathological hallmark of AD stimulated by synaptic activity. Production of the short Aβ peptides is not toxic, but aggregated Aβ fibrils are a pathological condition of AD (Soto et al., 2007). Although early studies have suggested that high Aβ accumulation is the cause of plaque formation and neuronal cell toxicity, they have concluded that even the short insoluble Aβ peptides may be more toxic (Coman & Nemeş, 2017). In a normal brain, an enzyme called α-secretase acts on APP and cleaves into secreted APPα (SAPPα) and an 83 amino acid membrane-bound C-terminal fragment called CTF83. Alternatively, in an AD brain, an enzyme called β-secretase acts on APP and cleaves it into secreted APPβ (SAPPβ) and a 99 amino acid long, membrane-bound C-terminal fragment called CTF99. In a normal brain, CTF83 is further cleaved by the γ-secretase complex and leads to the generation of APP intracellular domain (AICD) fragments, which translocate to the nucleus and regulate the proteins of neuroprotective pathways. However, in AD, γ-secretase cleaves the CTF99 fragment into Aβ40-42 peptides leading to the formation of Aβ fragments. Soluble Aβ fragments are dissolved whereas under certain conditions, insoluble Aβ fragments are aggregated leading to plaque formation and disruption in cell communication (Chaudhary et al., 2018; Du et al., 2018; Edwards, 2019; Octave, 1995) including abnormal deposit of amyloid β (Aβ. In humans, the estimated physiological rate of production of Aβ is 7.6% per hour and clearance rate is 8.3% per hour and proteolytic degradation is the major route of clearance. The targets particularly related to the formation of Aβ plaques have been mentioned below. Few targets directly contribute to Aβ plaques by cleaving APP directly (γ-secretase), whereas the others play their roles indirectly by over expression of apolipoprotein E (APOE), dual-specificity tyrosine-regulated kinases (DYRK1A), nod-like receptor protein 3 (NLRP3), casein kinase 1 (CK1), Cyclin-dependent kinase 5 (Cdk5), triggering receptor expressed on myeloid cells 2 (TREM2) and matrix metallopeptidases (MMP) as mentioned in Table 1.
β-secretase 1 (BACE1)
BACE1 was identified in 1999 and belongs to the class of aspartyl proteases with disulfide bond arrangement and consists of 501 amino acids. It is predominantly found in the regions of the brain and is said to be the major determinant of amyloidosis in the CNS (Evin & Hince, 2013). BACE1 is a key enzyme that regulates the β-secretase activity of APP. Mutation in APP leads to the increased access of the enzyme to β-secretase cleavage site and augments Aβ production (Vassar & Kandalepas, 2011). Deposition of Aβ peptides leads to the formation of large Aβ plaques in the brain and can distort the neuronal synapses. Lower Aβ levels can be achieved by decreasing BACE1, which may also reverse microglial activation and abnormality in neuronal functions (Coimbra et al., 2018; Yan et al., 1999). Accumulation of Aβ aggregates in the synapses affects synaptic plasticity leading to a decline in memory. BACE1-deficient mice exhibited cognitive impairments significantly. For this reason, BACE1 inhibition is logically beneficial for AD patients (Laird et al., 2005).
A reduction in Aβ deposition caused deletion of BACE1 in adult mice providing us with genetic evidence (Hu et al., 2018) which likely causes AD. Pharmacological inhibition of BACE1 may slow down the Aβ pathology, but to take advantage of this, treatment should be initiated before the spread of Aβ deposition. Therefore, inhibition of BACE1 may regulate to the extent that it prevents the formation of new plaques and delays the progression to AD (Blume et al., 2018; Das & Yan, 2017).
BACE1 is considered a potential target to treat AD, as it is a prime enzyme involved in the Aβ cascade as shown in Figure 2. Intense efforts are being made to find a drug that can inhibit BACE1, but because it plays an important role in various physiological processes, the challenge is to develop selective and specific BACE1 inhibitors (Cole & Vassar, 2007; Rajendran et al., 2008).
For over 20 years, studies have been done to design and develop a BACE1 inhibitor with good selectivity, bioavailability, and blood-brain barrier (BBB) permeability, but clinical trials indicate a high failure rate of many lead drug candidates. Many BACE1 inhibitors have been shelved due to adverse effects, and an inability to reduce AD-related symptoms even though plaque formation is reduced. Nevertheless, BACE1 inhibition is still considered the best target to stop AD, as it is the neck of the disease.
Verubecestat (MK-8931) is the first BACE1 inhibitor (a small molecule) from the Merck pharmaceutical Company with good bioavailability and BBB permeability. The administration of verubecestat to preclinical animals reduced the Aβ oligomers in cerebrospinal fluid (CSF) and the brain with no adverse effects. Phase 1 clinical trials also showed that it is well-tolerated and reduces the plaque concentration in the CSF. In phase 2 clinical trials, the focus was on testing the cognitive properties of the drug. Verubecestat did not exhibit any improvement in the cognitive capacity of AD participants and the clinical trial was terminated in February 2018. Side effects, like rashes, change in color, insomnia, and weight loss were reported by the participants (Egan et al., 2018; Kennedy et al., 2016; Scott et al., 2016; Zwan et al., 2017).
Lanabecestat, an oral BACE1 inhibitor developed by the AstraZeneca Pharmaceutical Company was tested first on primary cortical neurons of mice, guinea pigs, and dogs prior to clinical trials. A half-life of 9 hours means that it has a long-lasting reduction of Aβ oligomers. In 2014, in collaboration with Eli Lilly, phase I studies began, and the results were satisfactory with a good metabolic and tolerability profile in mild AD patients. Similar to Merck’s verubecestat, lanabecestat also decreased the levels of Aβ in CSF in the treated group. In phase 2 clinical trials that were conducted on over 1400 participants, its potency and safety were analyzed by amyloid positron emission tomography (PET) scans and the levels of Aβ in CSF were measured. The expected outcome of the study was an improvement in cognition and a decrease in Aβ levels in CSF. Unfortunately, the trials had to be discontinued as the lanabecestat treatment did not meet the primary endpoints (Cebers et al., 2017; Eketjäll et al., 2016; Sakamoto et al., 2017).
Atabecestat (JNJ-54861911) is another small-molecular BACE1 inhibitor developed by the Shionogi Pharmaceutical Company. This has entered the phase I clinical trial in collaboration with Janssen pharmaceuticals. The administration of atabecestat (5–150 mg) for 14 days showed a considerable reduction in Aβ levels in CSF and plasma. In another trial termed EARLY (NCT02569398) that was begun in 2015, the participants at risk of developing AD or suffering from mild AD were given the drug for 4.5 years with continuous monitoring of cognitive functions. Unfortunately, Janssen had to announce the termination of this trial as observations indicated a rise in the liver enzymes in a few patients. This suggested that it is associated with liver toxicity and the drug was discontinued on May 17, 2018 (Lahiri et al., 2014; Timmers et al., 2018).
Elenbecestat (E2609) was developed first by the Eisai Pharmaceutical Company in December 2014, as a small-molecule BACE1 inhibitor. After initial trials, a ninth phase 1 clinical trial was conducted in 2016, which was satisfactory at a dose of 50 mg in 16 healthy volunteers. In October 2016, a program called MISSION was initiated to examine the outcomes of PET and hippocampal Aβ volume changes. In 2018, Eisai planned for phase 2 clinical trials as it was found to be effective in reducing the brain Aβ levels and cognitive function. However, in 2019, BIOGEN/Eisai announced the termination of MÏSSION trials due to an unfavorable risk-benefit ratio and cognitive improvement was also not significant. However, the only relief was no hepatotoxicity, which has been a feature in clinical trials of other drugs (Imbimbo & Watling, 2019; Panza et al., 2010).
A new small-molecule BACE1 inhibitor CNP520 (umibecestat) from the Novartis Pharmaceutical Company is being developed. In preclinical models, CNP520 has been shown to reduce Aβ generation and penetrate the BBB with a good selectivity toward the target. No phase 1 clinical trial results have been reported, but phase 2 trials were conducted by Amgen Company aimed at delaying the progression or onset of AD. In July 2019, the study ended prematurely, citing cognitive decline and weight loss in the treatment groups. Researchers continued to monitor the cognition capacity and the level of Aβ in the brain after the treatment was stopped, and they found the cognitive decline and the hippocampus volume loss were reversible (Neumann et al., 2018). Regardless of the failures in clinical trials, discovering a BACE1 inhibitor should not be stopped. Inhibition of BACE1 along with other targets could be a potential strategy and is likely to be more effective in AD. Nevertheless, a breakthrough in the drug discovery for BACE1 inhibition happened on June 7, 2021. The first FDA-approved drug for AD treatment is aducanumab developed by the Biogen and Eisai Company under an accelerated approval pathway (Schneider, 2020).
γ-secretase (Presenilin)1
It is an integral intermembrane protein with many subunits forming a protease complex and consists of four individual proteins: PSEN1, PSEN2, APH1, and Nicastrin. γ-secretase and its components are generally synthesized in the endoplasmic reticulum, but for maturation, it requires the coordination of the ER-Golgi circuit (Basi et al., 2010; Wolfe, 2014).
Presenilin mutations were first seen in connection with familial AD, and further research established that PSEN1 along with structurally close PSEN2 are the catalytic components of γ-secretase, which is involved in the cleavage of APP at varying lengths. In AD, APP cleavage by γ-secretase happens at the CTF99 fragment (a fragment of APP after cleavage by BACE1 enzyme) of BACE1, leading to the formation of Aβ plaques. The hypothesized reason behind the action of γ-secretase is after the cleavage of α- or β-secretase into the large ectodomain of the substrate, which prevents binding to γ-secretase. As the final complex in APP processing, γ-secretase might be contributing to the levels of the Aβ and Notch signaling pathway as shown in Figure 3. Notch receptor is responsible for cell differentiation events in all multicellular organisms, in embryogenesis and adulthood (Basi et al., 2010; Bavishi et al., 2015).
The inhibition of γ-secretase presents an obvious door to block the production of Aβ (De Strooper, 2007; Wolfe, 2014). The first reported γ-secretase inhibitors (GSIs) were peptide aldehyde type calpain and proteasome inhibitors. Despite their less potency and lack of selectivity, these are said to be the answers for GSIs (Wolfe, 2012).
The complete inhibition of γ-secretase seemed a good approach. However, it was found that it has an extensive biological role and cleaves multiple proteins to yield physiologically essential products. The total inhibition of γ-secretase has resulted in severe adverse effects in vivo. This has been evident from the clinical trial conducted by Eli Lilly for the γ-secretase inhibitor called semagacestat. Patients experienced severe side effects, which led to the discontinuation of the clinical trial in 2010 (Kounnas et al., 2010; Schiering et al., 2016).
A substantial amount of research is being done for the discovery and development of small-molecule inhibitors for presenilin-containing γ-secretase complex as a potential therapy for AD (Kounnas et al., 2010).
The future focus will be developing an inhibitor that can reduce the Aβ levels without being involved in the Notch signaling pathway and discovering γ-secretase modulators, which can shift the Aβ1-42 to less pathogenic forms. Pharmacological inhibition of γ-secretase is a well-documented target for lowering plasma Aβ levels and has a greater impact on the pathology of AD (De Strooper et al., 2012; Wolfe, 2014).
PF-3084014, as a γ-secretase inhibitor, was in development in Pfizer but has to be abandoned due to the lack of data on its effect on cognition and the amount of Aβ plaques in animal models of AD (Panza et al., 2010).
GDI-953 a potent γ-secretase inhibitor developed by Wyeth has been shown to inhibit Aβ production at nanomolar concentrations in preclinical animals but in phase 2 clinical trials, it did not show much effect on the AD patients. The dose administered was not sufficient to clear the plaques or improve the cognition capacity of AD patients (Martone et al., 2009).
E-2012, a novel γ-secretase inhibitor reduces Aβ levels by modulating γ-secretase without interfering with the Notch processing. This drug was developed by Eisai in collaboration with torrey-pines therapeutics. In 2006, the drug candidate entered into phase 1 clinical but was terminated because of the side effects observed in preclinical animals after 13 weeks. The drug is now back and the clinical trial is under process and is in phase 1 (Panza et al., 2010).
FRM-36143 is another novel γ-secretase inhibitor that reverses the PSEN mutations without interfering with the Notch signaling pathway in the preclinical studies. Thus, FRM-36143 is a potential candidate for further research to take into the clinical sector (Blain et al., 2016).
As γ-secretases play multiple roles, targeting PSEN1 mutations specifically will be the best way to minimize and prevent disturbances in the physiological functions of the body.
Cdk5
Two decades have passed since the discovery of CDK. Cdk5 belongs to the family of cyclin-dependent kinases, which are necessary for brain development during embryogenesis and essential for various neuronal functions related to cognition and memory in adults. Cdk5 is mainly found to be active in post-mitotic neurons (Dhavan & Tsai, 2001).
In a normal brain, the activity of Cdk5 is highly regulated by specific cyclin-related molecules p35 and p39. Among the two, p35 is the most studied activator protein of Cdk5. Regulated Cdk5 maintains synaptic plasticity, the survival of neurons, and cognition. When the activity of Cdk5 is deregulated, it promotes oxidative stress and mitochondrial dysfunction (Shukla et al., 2012; Wang et al., 2015; Wilkaniec et al., 2018).
In case of stress, there is an increase in the level of calcium in the cytoplasm, leading to proteolytic cleavage by calpain. Calpain belongs to the family of calcium-dependent cysteine proteases that are involved in cognition and cell development, motility, apoptosis, and differentiation. Calpain has two forms associated with it m-calpain and μ-calpain. They require only milli molar and micro molar concentrations of calcium for their activation. Calpain cleaves p35 and p39 resulting in p25 and p29 as cleaved fragments. This p25 can activate Cdk5 leading to a higher and active Cdk5/p25 stable complex. This causes further Aβ formation, mitochondrial dysfunction, and other pathological events leading to degenerated neurons and apoptosis as represented in Figure 4 (Cruz & Tsai, 2004; Liu et al., 2003). In AD, plaques are formed due to the deposition of Aβ aggregates extracellularly. Over-expression of Cdk5 promotes further Aβ deposition resulting in neurotoxicity, activation of kinases, and further neurofibrillary tangles (NFT) formation (Liu et al., 2003; Reinhardt et al., 2019; Taniguchi et al., 2001).
One study showed that upon neurotoxicity by glutamate or Aβ, neurons up-regulated Cdk5 activity, but upon treatment with known Cdk5 inhibitors, cells showed a reduction of hyperactive Cdk5. This strongly indicates that hyperactivation of Cdk5 may be involved with phosphorylation of APP leading to excessive Aβ formation (Cruz & Tsai, 2004; Fischer et al., 2002; Shah & Lahiri, 2014). An increase in Cdk5 activity of p25 is observed in the post-mortem brains of AD patients making it a therapeutic target for AD.
In research related to Cdk5 inhibition, two inhibitors have been tested: Cdk5 inhibitor (roscovitine) (Huber & O’Day, 2012; Menn et al., 2010) and calpain inhibitor (MDL28170) for AD. Roscovitine is in phase 2 clinical trials for the treatment of lung and nasopharyngeal cancer. Cdk5 deregulation is related to melanoma and neurodegenerative diseases, suggesting that roscovitine can be further investigated for AD. Roscovitine exhibits neuroprotective activity in vitro and in vivo (Dhavan & Tsai, 2001; Khalil et al., 2015; Shah & Lahiri, 2014).
Studies on AD mouse models suggest that the inhibition of Cdk5 activity genetically or pharmacologically will prevent synaptic dysfunction and neuronal death (Seo et al., 2017). Trials with Cdk5 inhibitors in the clinical sector have not been promising due to side effects or off-target events (Cicenas et al., 2015).
AK275 is the first exogenous calpain inhibitor and was mostly preferred because of its selectivity, membrane permeability, solubility, and in vitro efficiency. AK275 is considered to be a neuroprotectant for the treatment of focal brain ischemia, indicating its potential for treating AD (Yildiz-Unal et al., 2015). There are other inhibitors that researchers are working on to improve their stability, potency, and efficiency with improved pharmacological profiles (Bartus et al., 1994; Wilkaniec et al., 2016).
In a recent study on mouse models of AD, anti-diabetic drugs troglitazone and pioglitazone belonging to the class of thiazolidinedones (TZD) have been shown to inhibit Cdk5 kinase activity and reduce synaptic deficits (Cho et al., 2013). However, the use of TZDs is more than usually related to edema, weight gain, and heart failure (Rizos et al., 2009), which raises a concern.
In another study on APP/PS1 mice, in which Cdk5 was hyperactivated in the hippocampus, an anti-diabetic drug metformin was administered. The reports showed that after chronic administration of metformin, the cleavage of p35 into p25 was prevented and in turn, inhibited Cdk5, which was assessed by a combination of pharmacological and molecular techniques. Also, the reports have shown that chronic administration of metformin restored impaired synaptic plasticity and improved cognitive function in mice (Wang et al., 2020). Collectively, this information suggests a captivating note that anti-diabetic drugs can be of use as drugs to treat neurodegenerative diseases.
In the end, drug candidates that can reduce the hyperactivity of Cdk5 may be regarded as a highly potential candidate to decrease the production of Aβ (Fischer et al., 2002).
Dual-specificity tyrosine-regulated kinases (DYRK1A)
DYRK1A is a dual specific tyrosine-phosphorylated regulated kinase enzyme that belongs to the DYRK family and is one among five protein kinases that direct signals in the development of the nervous system (Ferrer et al., 2005; Yang et al., 2001). DYRK1A is extensively found in the cerebellum, olfactory bulb region, and hippocampus, and is also present in the Down syndrome region on chromosome 21, and controls brain growth through neuronal proliferation and neurogenesis. DYRK1A is involved in the formation of Aβ plaques and NFT, which has been evident from the reports of higher concentrations of DYRK1A in the brains of AD patients (Souchet et al., 2019; Stotani et al., 2016). Although DYRK1 plays a role in the formation of plaques, it is essential for the brain during embryonic development in the early stages, where DYRK1 is involved in signaling pathways related to cell growth and proliferation, separation of cells into mature neurons, as well as in the formation of dendritic spines, which are important for the transmission of impulses. However, in a mature brain, DYRK1A can become aggressive and may initiate AD pathology (Galceran et al., 2003). The dysfunction of DYRK1A is a prominent feature of Down syndrome and patients with this disorder are highly susceptible to developing AD early in life (Velazquez et al., 2019).
When DYRK1 comes across APP, it binds to an aggregate of oxygen and phosphorus atoms leading to phosphorylation. High phosphorylation of these proteins leads to harmful effects in the brain. This hyperphosphorylation of APP leads to the formation of Aβ plaques as shown in Figure 5. Experimental evidence shows that the over-expression of DYRK1A contributes to enhanced β-amyloidosis and neurofibrillary degeneration. The over-expression of DYRK1A has been shown to increase the chances of proteolytic cleavage of APP and hence, an increased production of Aβ peptides. Besides, DYRK1A phosphorylates presenilin 1 (PS 1) and thereby increases γ-secretase activity causing more Aβ production (Souchet et al., 2019; Yang et al., 2001).
A drug repurposing study of CX-4945, a CK-2 inhibitor undergoing clinical trials for various cancers, shows that it inhibits DYRK1A-related AD pathology in mouse models (Chon et al., 2015). The inhibition of DYRK1A in chronic conditions has reversed memory problems in 3x-Tg AD mice. The results obtained were found to be associated with Aβ and tau pathology. It showed that the inhibition of DYRK1A reduced the Aβ plaque formation by inhibiting APP cleavage and also reduced insoluble tau phosphorylation (Branca et al., 2017; Pathak et al., 2018).
This suggests that drugs that can inhibit DYRK1A could be a possible therapeutic choice for AD. Knowledge of the mechanisms initiated by DYRK1A over-expression will get us closer to comprehending AD and developing effective therapies.
Some DYRK1A inhibitors have been tested in vitro but only a few have shown the potential to be clinical drug candidates. Harmine is an extensively studied DYRK1A inhibitor in preclinical research but because of its selectivity toward kinases, is not suitable for clinical development (Melchior et al., 2019).
ManRos Therapeutics is developing a small-molecule DYRK1A inhibitor named leucettine L41 based on the analog of leucettamine B derived from marine sponges (Burgy et al., 2013).
Samumed is developing a DYRK1A inhibitor named SM07883, which has been shown to protect against the hyperphosphorylation of tau and is currently under test in a phase 1 clinical trial (Melchior et al., 2019).
KVN93, a molecule that has considerably reduced DYRK1A kinase activity in vitro, is speculated to be a potential novel DYRK1A inhibitor that can improve cognitive deficits and regulate Aβ pathology (Lee et al., 2020).
Using the natural product epigallocatechin-3-gallate, the inhibition of excess DYRK1A activity and promoted improved cognitive functions were observed. However, these drugs are non-selective to the target, and possessing numerous off-target reactions has raised questions on their utility (Feki & Hibaoui, 2018).
Preclinical research shows that DYRK1A inhibitors are effective in slowing down the onset of AD in people suffering from Down’s syndrome. Despite many efforts to develop DYRK1A selective inhibitors, only a few of them have crossed the preclinical stage and their clinical manifestation remains to be tested further. The safety parameters are the primary concern for DYRK1A inhibitors and this depends on their kinase selectivity and potency toward the target.
Apolipoprotein E (APOE)
APOE is a gene present on chromosome 19 and makes a protein APOE that helps in the metabolism of fats in the body (Liu et al., 2013; Yamazaki et al., 2019). APOE has two regions an N-terminal, which is the receptor-binding region, and a C-terminal domain, which is a fat-binding region. The APOE gene in humans is classified into three polymorphic alleles (E2, E3, and E4) with a prevalence rate of 8.4%, 77.9%, and 13.7%, around the world. However, the frequency of the E4 allele is remarkably more up to 40% in AD patients. APOE consists of 299 amino acids with a molecular weight of 34 kDa. The number of amino acid residues differentiates the three APOE isoforms (Morris et al., 2010). An in vitro study in 1996 showed that APOE is involved in Aβ formation and the APOE4 allele is considered to be the driving factor over the E2 or E3 isoform (Adalbert et al., 2007). APOE is mainly found in the liver and brain and helps in transporting lipids among cells or tissues by binding to lipoprotein via the APOE4 receptor in the liver. ApoE4 slows down the rate of transportation of cholesterol from the blood, which may lead to coronary heart disease. In the brain, APOE is mainly secreted in the astrocytes. Various experiments have indicated that APOE isoforms can efflux cholesterol. APOE2 and APOE3 have a better efficiency than APOE4 in reducing the cholesterol levels. Cholesterol is needed for different functions of neurons, which include synaptic plasticity and maintaining dendritic spine density. The less ability of APOE4 in lipid metabolism affects the condition of a neuron leading to neuronal death as seen in Figure 6. Carriers of APOE4 tend to have enhanced pathology related to AD before clinical symptoms become evident. APOE4 also presents the pathogenesis of AD by independent Aβ mechanisms, which include changes in the synaptic plasticity, lipid homeostasis, and neuronal inflammation. The precise pathophysiology between APOE isoforms and Aβ metabolism is still unclear. Current studies have found out that APOE4 could alter the production, aggregation, and clearance of Aβ (Namba & Ikeda, 1991; Safieh et al., 2019). Furthermore, APOE4 carriers have lower levels of CSF Aβ42 and show higher PiB and PET binding than non-carriers (Liu et al., 2013). Moreover, APOE4 is believed to enhance Aβ production by affecting the activity of γ-secretase (Adalbert et al., 2007; Liu et al., 2017).
APOE2 is considered to be a protective allele against AD. APOE2 could delay AD by an average of 7.5-8.5 years. Scientists have developed an Adeno-associated virus (AAV) type of particle that was able to reduce the Aβ load and bring back the cholesterol to normal levels in transgenic mice. Bexarotene as a retinoid x receptor (RXR) reversed both brain pathologies and cognitive dysfunctions that are driven by APOE4 and also induced compensation for the lipid deficiency of APOE4. This suggests that RXR agonists may be a way to counteract the pathological effects of APOE4 leading to AD (Boehm-Cagan & Michaelson, 2014).
A recent in silico study reported that epicatechin-gallate has a binding affinity toward APOE4 and can be a potential lead compound to inhibit the progression of AD (Bano et al., 2019). Thus, elucidation of the pathogenic link between APOE4 and memory functions is a considerable challenge. Inhibitors selectively targeting APOE4 or increasing the expression of APOE2 could bring assistance in the way of drug development for AD. Another strategy that can be implemented is to edit the gene from the E4 variant to the E3 variant.
Casein kinase 1 (CK1):
Exactly two decades ago, the regulation of casein kinase in AD was reported. Casein kinase 1 belongs to the family of protein kinases, which are mainly involved in maintaining circadian rhythms. Until now, seven different isoforms of CK1 have been characterized, of which CK1-δ and CK1ε are expressed in the brain (Rodrigues & Silva, 2017).
CK1-δ and CK1ε play an important role in maintaining homeostasis of the circadian rhythm (Figure 7).
In specific genetic ablation, CK1-δ changes the period of circadian rhythm and CK1ε may result in arrhythmicity. This is one of the reasons subjects with AD go to sleep later than the rest of the population. Disruption of the clock by CK1 hyperactivity and mutations in the period gene PER may be a causative factor in the pathogenesis that underlies AD as described in Figure 8. An in vitro study demonstrated that CK1ε can phosphorylate the Aβ precursor APP and both β- and γ-secretases that can cleave APP (Chauhan et al., 1993; Yasojima et al., 2000).
There is an involvement of CK1 activity in the regulation of APP cleavage (Flajolet et al., 2007). A higher association of CK1 is seen in neurodegenerative lesions in the final stages of neuronal death in the brain, which is the same with AD. The key enzymes that are involved in the cleavage of APP (β-secretase and γ-secretase) are also related to CK1 as a target to inhibit plaque formation. The brains of AD patients have 30 times more CK1 than that of a normal human brain (Adler et al., 2019; Chauhan et al., 1993).
A research study (2019) done on APP-PS1 mice showed that the inhibition of CK1 improves cognitive function and also reduces the load of Aβ in the brain. Another study reported that Aβ stimulates casein kinases along with other protein kinases, which may then initiate tau hyperphosphorylation leading to neurofibrillary tangles and ultimately neuronal death (Manakadan et al., 2015; Sundaram et al., 2019). Further characterization and identification of new inhibitors of CK1 that can regulate AD-regulated circadian shifts can be focussed as it is essential for the clearance of Aβ.
A small-molecule inhibitor -PF-670462- of CK1δ/ε is found to alter rhythmicity, improve cognitive functions, and reduce the Aβ load in APP-PS1 mice (Sundaram et al., 2019).
PF-05251749, a CK1δ and ε inhibitor, initially developed by Pfizer Company for AD, was tested for phase 1 safety and tolerability following preclinical studies considering circadian rhythm as a major parameter for assessment. No further development has been updated since October 2018; however, Biogen acquired the molecule for further studies in the AD population in January 2020 (Benn & Dawson, 2020). Future research should concentrate on early intervention to reduce CK1ε/δ by understanding the prophylactic potential.
Nod-like receptor protein 3 (NLRP3)
NLRP3 is an intracellular oligomeric multi-protein complex that is being extensively studied for neurodegenerative and immune system-related diseases (Song et al., 2017). NLRP3 is also called an inflammasome that belongs to the family of nod-like receptor (NLR) and is present in astrocytes and microglia in the CNS and elicits an immune response against damaged signals. The NLRP3 protein complex consists of the sensor protein NLRP3, the adaptor protein, a caspase activating and recruiting domain, and pro-caspase-1. The assembly of this complex and its activation leads to the separation of procaspase-1 into active caspase-1. The activated caspase-1 in turn cleaves and activates proinflammatory cytokines. In AD, the aggregation of misfolded proteins or Aβ plaques triggers NLRP3 can act as a platform for the activation of caspase-1 that initiates an inflammatory response, which triggers the pro-inflammatory cytokines IL-1β and IL-18 that are released into the extracellular space. These promote various inflammatory and immune diseases and mediate neuronal death (Halle et al., 2008; Yang et al., 2018; Yang et al., 2019, Tan et al., 2013). The inflammasome constituents, such as NLRP1, NLRP3, ASC, and Caspase-1 along with the downstream effectors, like IL-1β and IL-18 are up-regulated at both mRNA and protein levels in AD patients Figure 8 describes the pathway of Aβ plaque formation by NLRP3.
A study conducted in 2013 assessed the amount of cleaved caspase-1 in AD patients and found that its levels were more compared to the controls. The same case was mirrored in aged APP/PS1 transgenic mice (Yin et al., 2018).
A study of a rationally designed NLRP3 inflammasome inhibitor (JC-124) in Transgenic CRND8 APP mice showed that upon treatment with (JC-124), the levels of Aβ deposition in the brains of CRND8 mice were reduced along with reduced APP cleavage (Heneka et al., 2013). This suggests that discovering agents that can control the activation of NLRP3 might be a potential target to tackle neuroinflammation and plaque formation.
Since NLRP3 inflammasome activation is a multistep process, inhibiting the activation of NLRP3 can be done by different approaches: Subduing molecules that initiate the NLRP3 complex formation, suppressing the upstream signals by inhibiting the NLRP3 directly or indirectly depending on the molecule targeted and inhibiting caspase-1 cleavage. Several molecules have been shown to inhibit NLRP activation and are said to have been validated in animal models. An in vivo study described that a potent small-molecule inhibitor MCC950 blocked NLRP3 activation selectively at nanomolar concentration (Coll et al., 2015). A recent study on MCC950 also demonstrated that it can block nigericin-induced activation of NLRP3 inflammasome by inhibiting efflux of chloride that acts as an upstream pathway for NLRP3 activation (Zahid et al., 2019). Cystic fibrosis transmembrane conductance regulator (CFTR) channel inhibitor C172 can also inhibit NLRP3 activation. Another report on C172-CY09 confirms that it selectively binds to NLRP3 inflammasome but not to any other and inhibits its ATPase activity. The ATPase activity of NLRP3 is important for the activation of NLRP3 and its oligomerization (Jiang et al., 2017).
Most predominantly, CY-09 showed an inhibitory effect on NLRP3 and good pharmacokinetic properties in terms of tolerability and bioavailability in a mouse model related to NLRP3-related disease. CY-09 is the first compound identified that was specifically able to inhibit NLRP3 both in vitro and in vivo. However, additional studies are needed for its selectivity towards NLRP3 (Jiang et al., 2017; Lamkanfi & Dixit, 2017).
OLT1177, which was identified as a candidate for the treatment of degenerative arthritis, has successfully cleared the phase I clinical trial, and now is in the phase 2 clinical trial for the treatment of acute gouty arthritis. Marchetti et al. described that OLT1177 possesses anti-inflammatory action and can inhibit NLRP3 inflammasome activation. Like CY-09, OLT1177 also directly binds to NLRP3 and inhibits ATPase activity (Marchetti et al., 2018; Toldo et al., 2019).
To date, many molecules have been identified and investigated as NLRP3 inflammasome inhibitors in mouse models, but only a few have clinical value. However, a new study identified that the anti-allergic drug, tranilast can be used to treat inflammatory diseases. Huang et al. reported that tranilast can inhibit NLRP3 specifically. Like the other specific inhibitors, tranilast does not interfere with the signaling pathways of NLRP3 inflammasome. Tranilast can also inhibit NLRP3 inflammasome activation in an ATPase-independent manner. Further, in vivo studies have shown that tranilast has a preventive effect on mouse models related to NLRP3-related diseases. The safety profile needs to be investigated further (Feng et al., 2020; Huang et al., 2018).
A widely used OTC herbal medicine for the treatment of inflammatory diseases, Rabdosia Rubescens, has an active constituent, oridonin, which is reported to exhibit anti-inflammatory effects (Kuo et al., 2014). Previous reports have mentioned that oridonin can suppress inflammatory mediators and exhibit therapeutic effects on neuroinflammation. Studies have suggested that oridonin can specifically inhibit NLRP3 inflammasome activation (Kuo et al., 2014; Wang et al., 2019; Wang et al., 2015; Yang et al., 2019). It is used in clinical practices for different diseases but studies need to be further done for its therapeutic potential in NLRP3 inhibition associated with AD.
Matrix metallopeptidases (MMP)
Matrix metallopeptidases are also known as matrixins or matrix metalloproteinases and are calcium-dependent proteases belonging to the metzincin superfamily. The MMP family consists of other 28 MMPs, which are enzymes capable of degrading and processing many proteins in the extracellular matrix. MMPs are further divided into six main subgroups, of which gelatinase type (MMP-2 and 9) and membrane-type MMPs (MMP-1, 2, 3, 4, 5, and 6) play a role in AD and other neurological diseases (Wang et al., 2014; Yin et al., 2006). MMPs play an important role in cytokine inactivation responsible for inflammation, and physiological processes, such as neurogenesis and angiogenesis. MMPS is generally expressed in neurons but is also found in astrocytes and microglia. Astrocytes have been recently considered mediators in the degradation of Aβ. Evidence suggests that MMP-2, MMP-3, and MMP-9 are important players in AD compared to the other MMPs (Brkic et al., 2015; Duits et al., 2015; Yin et al., 2006; Yoshiyama et al., 2000)
MMP-2 is the major MMP that is directly linked to Aβ in the brain and the dysfunction of this enzyme exerts influence on the processing of Aβ(1-40/42) in vitro and in vivo, as deletion of MMP-2 leads to a greater Aβ accumulation rather than MMP-9 knockout. The proteolytic activity of MMP-2 and MMP-9 in clinically diagnosed AD patients showed higher activity of MMP-9 but not MMP-2 when compared to healthy brains suggesting that MMP-2 may have a protective role in AD (Yin et al., 2006). Expression MMP-9 is seen in cytoplasm, NFT, senile plaques, hippocampus, and cerebral cortex in AD patients. A post-mortem report of human frontal and parietal cortical tissues obtained from clinically diagnosed AD patients has shown a higher activity of MMP-9 rather than MMP-2 compared to healthy brain samples. A recent study also found that astrocytes present near the Aβ plaques have shown an increased expression of MMP-9 in aged APP/PS1 mice (Brkic et al., 2015; Fragkouli et al., 2014). From these reports, a piece of conclusive evidence can be drawn that MMP-9 is over-expressed and is found to be higher in the plasma of people with AD. As AD is a complex disorder with many agents playing a role, targeting more than one MMP with pan-specific inhibitors or combination of MMP with another target of AD can also be explored (Lorenzl et al., 2003).
Minocycline, an anti-inflammatory drug that is used for multiple sclerosis and Huntington’s disease, was tested for its effect on AD. It showed good tolerability and pharmacokinetic profile in vitro and in vivo. However, during a randomized clinical trial conducted on AD patients, it did not show any improvement in the cognitive capacity nor did it slow down the progression of AD. The negativity is explained because of the poor dose that has been tested in animals. This can be further explored for AD by altering the doses in the preclinical studies and thereby testing in humans considering that minocycline has a good penetration of BBB (Howard et al., 2020; Rosenberg et al., 2015; Yong et al., 2007).
MT-5-MMP is a protease that has come into the limelight as a potential target related to Aβ plaques in AD. A study on a 5xFAD mouse model of AD showed that MT5-MMP deficiency reduced Aβ levels in the cortex and hippocampus suggesting that inhibitors targeting MMP-5 specifically can be explored for AD (Baranger et al., 2016).
ZHAWOC7726, a TIMP peptidomimetic protein, has a good selectivity toward MMP-9, MMP-12, and MMP-13 for the treatment of cancer. This molecule could be further studied as it acts on different MMPs associated with AD (Gall et al., 2019).
Neprilysin (NEP)
Neprilysin, also called membrane metalloendopeptidase (MME), is an enzyme encoded by the MME gene in humans and cleaves peptides at the amino side of hydrophobic residues (Vodovar et al., 2015). It is found in a variety of tissues but more prominently is expressed in the kidneys and to a lesser amount in the brain and is an enzyme that controls Aβ degradation. NEP has a higher affinity toward the Aβ than other neuropeptides. Figure 9 shows the role of neprilysin in the degradation of Aβ plaques (Marr & Hafez, 2014; Webster et al., 2014).
During the early stages of AD, NEP is inactivated and down-regulated. Early studies have suggested that maintaining NEP levels in the brain could be a potential therapeutic strategy to slow the progression of AD (El-Amouri et al., 2008; Huang et al., 2006; Madani et al., 2006). The importance of NEP in Aβ regulation was demonstrated by using NEP knockout experiments. Elevated levels of Aβ species were seen in the hippocampus and brainstem when mice deficient for the NEP2 gene were used. Increased Aβ levels were observed in NEP2 knockout mice mated with APP Tg mice (El-Amouri et al., 2008; Yasojima et al., 2000).
The discovery of endopeptidases, such as NEP and NEP2 has opened up doors for viral-mediated gene therapy. While NEP has been a prominent target, NEP2 also can be considered selectively for Aβ clearance. A study has reported that the destruction of NEP has increased the levels of Aβ in the brain of mice (Huang et al., 2006; Madani et al., 2006; Maruyama et al., 2005).
Taking into account that NEP expression and activity are reduced in AD, the idea of increasing the NEP up-regulation might be beneficial. Saito et al. reported that somatostatin, a neuropeptide, and a NEP substrate can up-regulate NEP activity by regulatory the feedback cycle. Activation of somatostatin receptor subtype 4 increases the activity of NEP in the cortical regions suggesting that somatostatin can be considered an interesting target for increasing NEP levels (Bavishi et al., 2015; Eckman & Eckman, 2005). Hence, effective strategies and studying the pathways to increase the levels of NEP expression and activity may give us new opportunities for therapeutic interventions in AD as NEP inhibition has been a successful therapeutic candidate in heart failure with less ejection fraction (Pavo et al., 2020).
Dopamine 2 receptor (D2R)
Dopamine is a neurotransmitter and is generally expressed in the limbic system and cortex, which is related to mood and emotional stability (Ambrée et al., 2009; Kemppainen et al., 2003). Dopamine generally acts through five different receptors (D1, D2, D3, D4, and D5). Cortical functions are influenced by the number of dopamine (D2) receptors available. AD is associated with deficits in several neurotransmitters but cholinergic and dopamine systems are the most investigated in neuronal research. The levels of dopamine were found to be higher in AD patients than in controls (Martorana & Koch, 2014; Reeves et al., 2017).
Dopamine-containing neurons are mainly located in the midbrain. Dopamine plays an important role in synaptic transmission. Being a neuromodulator, there are chances of dopamine affecting the neurotransmitter release and membrane excitability of the pre and post-synaptic cells (Nobili et al., 2017). Dopamine can modulate the activity of cholinergic release from the neurons located in the basal forebrain. The dysfunction of dopamine is being seen as a new player in the pathogenesis of AD (Liu et al., 2016).
A 1986 report showed that the administration of levodopa restored cholinergic neurotransmission (Rinne et al., 1986). Dopamine regulates synaptic plasticity and also can maintain cognitive functions. Maintaining dopamine levels may reduce plaque formation by increasing synaptic plasticity and reducing oxidative stress (Cross et al., 1981; Martorana & Koch, 2014; Pan et al., 2019).
A recent study showed a link between the ventral tegmental area (VTA) and other parts of the brain. They analyzed the subjects by using 3-Tesla MRI technology. The reports indicated that there is a relationship between size and the functionality of VTA, the size of the hippocampus, and learning ability. The smaller size of VTA indicated that only a lesser amount of dopamine goes to the hippocampus and reduced memory performance. They concluded that this might be the reason for AD patients have memory loss, suggesting agents that can increase the dopamine levels in the body can reduce plaque formation and the progression of AD (Pan et al., 2019). Thus, maintaining the dopamine levels in the brain can at least delay the progression of AD by stabilizing the signaling pathway and reducing neuronal degradation.
Triggering receptor expressed on myeloid cells 2 (TREM2)
In the CNS, TREM2 is expressed mainly in microglia and is involved in the production of proinflammatory cytokines and controls neuronal inflammatory events as depicted in Figure 10 (Gratuze et al., 2018; Karanfilian et al., 2020). Being a transmembrane receptor, the involvement of immune and inflammatory pathways supports the fact that it has a role in AD. TREM2 is necessary for maintaining the microglial progression to fully mature disease-associated microglia (DAM). The expression of TREM2 is associated with plaque formation. Microglia respond to Aβ accumulation and become DAM (Jay et al., 2017).
Microglia have been shown to surround and limit Aβ plaques in the AD brain in the absence of microglial scavenging receptors and resulted in decreased clearance of Aβ by microglia (Carmona et al., 2018; Jay et al., 2017; Zheng et al., 2018). So far, 46 variants of TREM2 have been worked upon about AD. A rare genetic variant p. Arg47His (rs 75932628) is said to have an increased risk of developing AD in the European and American populations. However, the association of the same has not been prominent in Asian countries, suggesting that TREM2 may play a population-specific role (Jay et al., 2017).
Several in vitro and in vivo studies have been done concerning TREM2 and Aβ burden. In vitro studies have shown that TREM2 is strongly involved in Aβ40 and Aβ42 uptake by the microglial cells and plaque formation. In vivo studies conducted on different mouse models suggest an age-dependent effect on Aβ deposition (Cheng et al., 2016).
TREM2 may result in greater Aβ deposition in the early stages of the disease and then becomes beneficial in the end stages by interfering with inflammatory signaling and maintaining the ability of microglia to recover the injured neuron (Carmona et al., 2018). A full understanding of the dual role of TREM2 in neurodegenerative disease, especially in AD is important. Addressing questions on whether altered phagocytosis of TREM2 is the reason for the accumulation of Aβ plaques or is it the interaction of TREM2 with other targets of AD can be a viable therapeutic strategy.
PY134 developed by Pionyr’s clinical programs for specifically targeting TREM2 in cancer is in phase 1 clinical trial. As the molecule can bind to TREM2 specifically, further studies can be done to explore its efficacy in AD (Tang et al., 2019).
Activating a particular enzyme or target is more difficult than inhibiting it. A study suggests that a lector antibody (AL002) can bind and activate TREM2. The mouse version of the antibody, AL002a, binds to and activates TREM2. In cell culture, AL002a treatment increased the phosphorylation of Syk, a downstream effector of TREM2 signaling. Another study has also reported that a similar monoclonal antibody developed in Germany is said to activate TREM2 signaling by boosting the phospho-Syk signaling. Anti-TREM2 antibodies could perhaps serve as biomarkers, as well as therapeutic agents (Wang et al., 2020).
3. Recent developments
June 7, 2021, can be termed a historic day in the therapeutic sector of AD as aducanumab, a monoclonal antibody was approved by the Food and Drug Administration (FDA) as a drug of choice to treat AD. The drug is branded as Aduhelm and is licensed by Biogen and Eisai pharmaceutical companies under collaborative development. The FDA has approved this therapy under the accelerated approval pathway to benefit patients with serious neurodegenerative diseases following the priority review. Aduhelm works by targeting the Aβ in the brain and reduces the amount of Aβ plaques, potentially slowing down the neurodegeneration and progression of the disease (Schneider, 2020; Sevigny et al., 2016).
4. Conclusion
Several studies support the amyloidal theory that Aβ is the starter to a complex number of pathological events in the brain concerning AD. Aβ starts to emerge long before the clinical symptoms appear physically. The deposition of Aβ not only causes hindrance to the signaling system but also triggers cerebral deficits, calcium homeostasis, and cognition problems. Unfortunately, the research on Aβ plaques as a target for AD has been on a downward graph because of the failure of drugs in clinical trials. Even so, the re-launch of aducanumab as an anti-amyloid drug has stirred up this theory once again. The difficulty in pacing up with molecular knowledge is still evident as we still depend heavily on classical pathology. However, studies are done at a faster pace to identify the exact pathology behind AD suggesting that it is feasible to provide new targets for drug discovery and development directed toward the Aβ accumulation. In this article, we listed down the targets that play various roles in plaque formation, which could help design effective drugs for AD.
The best possible way to eliminate Aβ pathology is to prevent it from accumulating in the brain. The outcome that occurs on removing these pathological lesions in AD patients should be intensely focussed in the research studies.
Ethical Considerations
Compliance with ethical guidelines
There were no ethical considerations to be considered in this research.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Authors' contributions
All authors equally contributed to preparing this article.
Conflict of interest
The authors declared no conflict of interest
References
Adalbert, R., Gilley, J., & Coleman, M. P. (2007). Abeta, tau and ApoE4 in Alzheimer's disease: The axonal connection. Trends in Molecular Medicine, 13(4), 135–142. [DOI:10.1016/j.molmed.2007.02.004] [PMID]
Adler, P., Mayne, J., Walker, K., Ning, Z., & Figeys, D. (2019). Therapeutic targeting of casein kinase 1δ/ε in an alzheimer's disease mouse model. Journal of Proteome Research, 18(9), 3383–3393. [DOI:10.1021/acs.jproteome.9b00312] [PMID]
Ambrée, O., Richter, H., Sachser, N., Lewejohann, L., Dere, E., & de Souza Silva, M. A., et al. (2009). Levodopa ameliorates learning and memory deficits in a murine model of Alzheimer's disease. Neurobiology of Aging, 30(8), 1192–1204. [DOI:10.1016/j.neurobiolaging.2007.11.010] [PMID]
Ashraf, G. M., Chibber, S., Mohammad, Zaidi, S. K., Tabrez, S., & Ahmad, A., et al. (2016). Recent updates on the association between alzheimer's disease and vascular dementia. Medicinal Chemistry, 12(3), 226–237. [DOI:10.2174/1573406411666151030111820] [PMID]
Bano, S., Rasheed, M. A., Jamil, F., Ibrahim, M., & Kanwal, S. (2019). In silico identification of novel apolipoprotein e4 inhibitor for alzheimer's disease therapy. Current Computer-Aided Drug Design, 15(1), 97–103. [DOI:10.2174/1573409914666181008164209] [PMID]
Baranger, K., Marchalant, Y., Bonnet, A. E., Crouzin, N., Carrete, A., & Paumier, J. M., et al. (2016). MT5-MMP is a new pro-amyloidogenic proteinase that promotes amyloid pathology and cognitive decline in a transgenic mouse model of Alzheimer's disease. Cellular and Molecular Life Sciences, 73(1), 217–236. [DOI:10.1007/s00018-015-1992-1] [PMID] [PMCID]
Bartus, R. T., Baker, K. L., Heiser, A. D., Sawyer, S. D., Dean, R. L., & Elliott, P. J., et al. (1994). Postischemic administration of AK275, a calpain inhibitor, provides substantial protection against focal ischemic brain damage. Journal of Cerebral Blood Flow and Metabolism, 14(4), 537–544. [DOI:10.1038/jcbfm.1994.67] [PMID]
Basi, G. S., Hemphill, S., Brigham, E. F., Liao, A., Aubele, D. L., & Baker, J., et al. (2010). Amyloid precursor protein selective gamma-secretase inhibitors for treatment of Alzheimer's disease. Alzheimer's Research & Therapy, 2(6), 36. [DOI:10.1186/alzrt60] [PMID] [PMCID]
Bavishi, C., Messerli, F. H., Kadosh, B., Ruilope, L. M., & Kario, K. (2015). Role of neprilysin inhibitor combinations in hypertension: Insights from hypertension and heart failure trials. European Heart Journal, 36(30), 1967–1973. [DOI:10.1093/eurheartj/ehv142] [PMID]
Benn, C. L., & Dawson, L. A. (2020). Clinically precedented protein kinases: Rationale for their use in neurodegenerative disease. Frontiers in Aging Neuroscience, 12, 242.[DOI:10.3389/fnagi.2020.00242] [PMID] [PMCID]
Benny, A., & Thomas, J. (2019). Essential oils as treatment strategy for alzheimer's disease: Current and future perspectives. Planta Medica, 85(3), 239–248. [DOI:10.1055/a-0758-0188] [PMID]
Blain, J. F., Bursavich, M. G., Freeman, E. A., Hrdlicka, L. A., Hodgdon, H. E., & Chen, T., et al. (2016). Characterization of FRM-36143 as a new γ-secretase modulator for the potential treatment of familial Alzheimer's disease. Alzheimer's Research & Therapy, 8(1), 34. [DOI:10.1186/s13195-016-0199-5] [PMID] [PMCID]
Blume, T., Filser, S., Jaworska, A., Blain, J. F., Koenig, G., & Moschke, K., et al. (2018). BACE1 inhibitor MK-8931 alters formation but not stability of dendritic spines. Frontiers in Aging Neuroscience, 10, 229. [DOI:10.3389/fnagi.2018.00229] [PMID] [PMCID]
Boehm-Cagan, A., & Michaelson, D. M. (2014). Reversal of apoE4-driven brain pathology and behavioral deficits by bexarotene. The Journal of Neuroscience, 34(21), 7293–7301. [DOI:10.1523/JNEUROSCI.5198-13.2014] [PMID] [PMCID]
Branca, C., Shaw, D. M., Belfiore, R., Gokhale, V., Shaw, A. Y., & Foley, C., et al. (2017). Dyrk1 inhibition improves Alzheimer's disease-like pathology. Aging Cell, 16(5), 1146–1154. [DOI:10.1111/acel.12648] [PMID] [PMCID]
Brkic, M., Balusu, S., Libert, C., & Vandenbroucke, R. E. (2015). Friends or foes: Matrix metalloproteinases and their multifaceted roles in neurodegenerative diseases. Mediators of Inflammation, 2015, 620581. [DOI:10.1155/2015/620581] [PMID] [PMCID]
Burgy, G., Tahtouh, T., Durieu, E., Foll-Josselin, B., Limanton, E., & Meijer, L., et al. (2013). Chemical synthesis and biological validation of immobilized protein kinase inhibitory Leucettines. European Journal of Medicinal Chemistry, 62, 728–737. [DOI:10.1016/j.ejmech.2013.01.035] [PMID]
Carmona, S., Zahs, K., Wu, E., Dakin, K., Bras, J., & Guerreiro, R. (2018). The role of TREM2 in alzheimer's disease and other neurodegenerative disorders. The Lancet. Neurology, 17(8), 721–730. [DOI:10.1016/S1474-4422(18)30232-1] [PMID]
Cebers, G., Alexander, R. C., Haeberlein, S. B., Han, D., Goldwater, R., & Ereshefsky, L., et al. (2017). AZD3293: Pharmacokinetic and pharmacodynamic effects in healthy subjects and patients with alzheimer's disease. Journal of Alzheimer's Disease, 55(3), 1039–1053. [DOI:10.3233/JAD-160701] [PMID]
Chaudhary, A., Maurya, P. K., Yadav, B. S., Singh, S., & Mani, A. (2018). Current therapeutic targets for alzheimer’s disease. Journal of Biomedicine, 3, 74-84. [DOI:10.7150/jbm.26783]
Chauhan, A., Chauhan, V. P., Murakami, N., Brockerhoff, H., & Wisniewski, H. M. (1993). Amyloid beta-protein stimulates casein kinase I and casein kinase II activities. Brain Research, 629(1), 47–52. [DOI:10.1016/0006-8993(93)90479-7] [PMID]
Cheng, J., Guo, X., Zhang, T., Zhong, L., Bu, G., & Chen, X. (2016). TREMs in alzheimer's disease: Genetic and clinical investigations. Clinica Chimica Acta; International Journal of Clinical Chemistry, 463, 88–95. [DOI:10.1016/j.cca.2016.10.022] [PMID]
Cho, D. H., Lee, E. J., Kwon, K. J., Shin, C. Y., Song, K. H., & Park, J. H., et al. (2013). Troglitazone, a thiazolidinedione, decreases tau phosphorylation through the inhibition of cyclin-dependent kinase 5 activity in SH-SY5Y neuroblastoma cells and primary neurons. Journal of Neurochemistry, 126(5), 685–695. [DOI:10.1111/jnc.12264] [PMID]
Chon, H. J., Bae, K. J., Lee, Y., & Kim, J. (2015). The casein kinase 2 inhibitor, CX-4945, as an anti-cancer drug in treatment of human hematological malignancies. Frontiers in Pharmacology, 6, 70. [DOI:10.3389/fphar.2015.00070] [PMID] [PMCID]
Cicenas, J., Kalyan, K., Sorokinas, A., Stankunas, E., Levy, J., & Meskinyte, I., et al. (2015). Roscovitine in cancer and other diseases. Annals of Translational Medicine, 3(10), 135. [DOI:10.3978/j.issn.2305-5839.2015.03.61]
Coimbra, J. R. M., Marques, D. F. F., Baptista, S. J., Pereira, C. M. F., Moreira, P. I., & Dinis, T. C. P., et al. (2018). Highlights in BACE1 inhibitors for alzheimer's disease treatment. Frontiers in Chemistry, 6, 178. [DOI:10.3389/fchem.2018.00178] [PMID] [PMCID]
Cole, S. L., & Vassar, R. (2007). The alzheimer's disease beta-secretase enzyme, BACE1. Molecular Neurodegeneration, 2, 22. [DOI:10.1186/1750-1326-2-22] [PMID] [PMCID]
Coll, R. C., Robertson, A. A., Chae, J. J., Higgins, S. C., Muñoz-Planillo, R., & Inserra, M. C., et al. (2015). A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nature Medicine, 21(3), 248–255. [DOI:10.1038/nm.3806] [PMID] [PMCID]
Coman, H., & Nemeş, B. (2017). New therapeutic targets in alzheimer’s disease. International Journal of Gerontology, 11(1), 2-6. [DOI:10.1016/j.ijge.2016.07.003]
Cross, A. J., Crow, T. J., Perry, E. K., Perry, R. H., Blessed, G., & Tomlinson, B. E. (1981). Reduced dopamine-beta-hydroxylase activity in Alzheimer's disease. British Medical Journal, 282(6258), 93–94. [DOI:10.1136/bmj.282.6258.93] [PMID] [PMCID]
Cruz, J. C., & Tsai, L. H. (2004). Cdk5 deregulation in the pathogenesis of Alzheimer's disease. Trends in Molecular Medicine, 10(9), 452–458. [DOI:10.1016/j.molmed.2004.07.001] [PMID]
Das, B., & Yan, R. (2017). Role of BACE1 in alzheimer's synaptic function. Translational Neurodegeneration, 6, 23. [DOI:10.1186/s40035-017-0093-5] [PMID] [PMCID]
De Strooper B. (2007). Loss-of-function presenilin mutations in Alzheimer disease. Talking point on the role of presenilin mutations in Alzheimer disease. EMBO Reports, 8(2), 141–146.[DOI:10.1038/sj.embor.7400897] [PMID] [PMCID]
De Strooper, B., Iwatsubo, T., & Wolfe, M. S. (2012). Presenilins and γ-secretase: Structure, function, and role in alzheimer disease. Cold Spring Harbor Perspectives in Medicine, 2(1), a006304. [DOI:10.1101/cshperspect.a006304] [PMID] [PMCID]
Dhavan, R., & Tsai, L. H. (2001). A decade of CDK5. Nature reviews. Molecular Cell Biology, 2(10), 749–759.[DOI:10.1038/35096019] [PMID]
Du, X., Wang, X., & Geng, M. (2018). Alzheimer's disease hypothesis and related therapies. Translational Neurodegeneration, 7, 2. [DOI:10.1186/s40035-018-0107-y] [PMID] [PMCID]
Duits, F. H., Hernandez-Guillamon, M., Montaner, J., Goos, J. D., Montañola, A., & Wattjes, M. P., et al. (2015). Matrix metalloproteinases in alzheimer's disease and concurrent cerebral microbleeds. Journal of Alzheimer's Disease, 48(3), 711–720. [DOI:10.3233/JAD-143186] [PMID]
Eckman, E. A., & Eckman, C. B. (2005). Abeta-degrading enzymes: Modulators of alzheimer's disease pathogenesis and targets for therapeutic intervention. Biochemical Society Transactions, 33(Pt 5), 1101–1105. [DOI:10.1042/BST20051101] [PMID]
Edwards F. A. (2019). A unifying hypothesis for alzheimer's disease: From plaques to neurodegeneration. Trends in Neurosciences, 42(5), 310–322. [DOI:10.1016/j.tins.2019.03.003] [PMID]
Egan, M. F., Kost, J., Tariot, P. N., Aisen, P. S., Cummings, J. L., & Vellas, B.,et al. (2018). Randomized trial of verubecestat for mild-to-moderate alzheimer's disease. The New England Journal of Medicine, 378(18), 1691–1703. [DOI:10.1056/NEJMoa1706441] [PMID] [PMCID]
Eketjäll, S., Janson, J., Kaspersson, K., Bogstedt, A., Jeppsson, F., & Fälting, J., et al. (2016). AZD3293: A novel, orally active bace1 inhibitor with high potency and permeability and markedly slow off-rate kinetics. Journal of Alzheimer's Disease, 50(4), 1109–1123. [DOI:10.3233/JAD-150834] [PMID] [PMCID]
El-Amouri, S. S., Zhu, H., Yu, J., Marr, R., Verma, I. M., & Kindy, M. S. (2008). Neprilysin: An enzyme candidate to slow the progression of Alzheimer's disease. The American Journal of Pathology, 172(5), 1342–1354. [DOI:10.2353/ajpath.2008.070620] [PMID] [PMCID]
Evin, G., & Hince, C. (2013). BACE1 as a therapeutic target in Alzheimer's disease: Rationale and current status. Drugs & Aging, 30(10), 755–764. [DOI:10.1007/s40266-013-0099-3] [PMID]
Feki, A., & Hibaoui, Y. (2018). DYRK1A protein, a promising therapeutic target to improve cognitive deficits in down syndrome. Brain Sciences, 8(10), 187. [DOI:10.3390/brainsci8100187] [PMID] [PMCID]
Feng, Y. S., Tan, Z. X., Wu, L. Y., Dong, F., & Zhang, F. (2020). The involvement of NLRP3 inflammasome in the treatment of Alzheimer's disease. Ageing Research Reviews, 64, 101192.[DOI:10.1016/j.arr.2020.101192] [PMID]
Ferrer, I., Barrachina, M., Puig, B., Martínez de Lagrán, M., Martí, E., & Avila, J., et al. (2005). Constitutive Dyrk1A is abnormally expressed in Alzheimer disease, Down syndrome, Pick disease, and related transgenic models. Neurobiology of Disease, 20(2), 392–400. [DOI:10.1016/j.nbd.2005.03.020] [PMID]
Fischer, A., Sananbenesi, F., Schrick, C., Spiess, J., & Radulovic, J. (2002). Cyclin-dependent kinase 5 is required for associative learning. The Journal of Neuroscience, 22(9), 3700–3707. [DOI:10.1523/JNEUROSCI.22-09-03700.2002] [PMID] [PMCID]
Flajolet, M., He, G., Heiman, M., Lin, A., Nairn, A. C., & Greengard, P. (2007). Regulation of alzheimer's disease amyloid-beta formation by casein kinase I. Proceedings of the National Academy of Sciences of the United States of America, 104(10), 4159–4164. [DOI:10.1073/pnas.0611236104] [PMID] [PMCID]
Fragkouli, A., Tsilibary, E. C., & Tzinia, A. K. (2014). Neuroprotective role of MMP-9 overexpression in the brain of alzheimer's 5xFAD mice. Neurobiology of Disease, 70, 179–189.[DOI:10.1016/j.nbd.2014.06.021] [PMID]
Galceran, J., de Graaf, K., Tejedor, F. J., & Becker, W. (2003). The MNB/DYRK1A protein kinase: Genetic and biochemical properties. Journal of Neural Transmission. Supplementum, (67), 139–148. [DOI:10.1007/978-3-7091-6721-2_12] [PMID]
Gall, F. M., Hohl, D., Frasson, D., Wermelinger, T., Mittl, P. R. E., & Sievers, M., et al. (2019). Drug design inspired by nature: Crystallographic detection of an auto-tailored protease inhibitor template. Angewandte Chemie, 58(12), 4051–4055. [DOI:10.1002/anie.201812348] [PMID]
Gopalakrishnan, A., Shankarappa, S. A., & Rajanikant, G. K. (2019). Hydrogel scaffolds: Towards restitution of ischemic stroke-injured brain. Translational Stroke Research, 10(1), 1–18. [DOI:10.1007/s12975-018-0655-6] [PMID]
Gratuze, M., Leyns, C. E. G., & Holtzman, D. M. (2018). New insights into the role of TREM2 in alzheimer's disease. Molecular Neurodegeneration, 13(1), 66. [DOI:10.1186/s13024-018-0298-9] [PMID] [PMCID]
Halle, A., Hornung, V., Petzold, G. C., Stewart, C. R., Monks, B. G., & Reinheckel, T., et al. (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nature Immunology, 9(8), 857–865. [DOI:10.1038/ni.1636] [PMID] [PMCID]
Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., & Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature, 493(7434), 674–678. [DOI:10.1038/nature11729] [PMID] [PMCID]
Howard, R., Zubko, O., Bradley, R., Harper, E., Pank, L., & O'Brien, J., et al. (2020). Minocycline at 2 different dosages vs placebo for patients with mild alzheimer disease: A randomized clinical trial. JAMA Neurology, 77(2), 164–174. [DOI:10.1001/jamaneurol.2019.3762] [PMID] [PMCID]
Hu, X., Das, B., Hou, H., He, W., & Yan, R. (2018). BACE1 deletion in the adult mouse reverses preformed amyloid deposition and improves cognitive functions. The Journal of Experimental Medicine, 215(3), 927–940. [DOI:10.1084/jem.20171831] [PMID] [PMCID]
Huang, S. M., Mouri, A., Kokubo, H., Nakajima, R., Suemoto, T., & Higuchi, M.,et al. (2006). Neprilysin-sensitive synapse-associated amyloid-beta peptide oligomers impair neuronal plasticity and cognitive function. The Journal of Biological Chemistry, 281(26), 17941–17951. [DOI:10.1074/jbc.M601372200] [PMID]
Huang, Y., Jiang, H., Chen, Y., Wang, X., Yang, Y., & Tao, J., et al. (2018). Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Molecular Medicine, 10(4), e8689. [DOI:10.15252/emmm.201708689] [PMID] [PMCID]
Huber, R. J., & O'Day, D. H. (2012). The cyclin-dependent kinase inhibitor roscovitine inhibits kinase activity, cell proliferation, multicellular development, and Cdk5 nuclear translocation in Dictyostelium discoideum. Journal of Cellular Biochemistry, 113(3), 868–876. [DOI:10.1002/jcb.23417] [PMID]
Imbimbo, B. P., & Watling, M. (2019). Investigational BACE inhibitors for the treatment of Alzheimer's disease. Expert Opinion On Investigational Drugs, 28(11), 967–975. [DOI:10.1080/13543784.2019.1683160] [PMID]
Jay, T. R., von Saucken, V. E., & Landreth, G. E. (2017). TREM2 in neurodegenerative diseases. Molecular Neurodegeneration, 12(1), 56. [DOI:10.1186/s13024-017-0197-5] [PMID] [PMCID]
Jiang, H., He, H., Chen, Y., Huang, W., Cheng, J., & Ye, J., et al. (2017). Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. The Journal of Experimental Medicine, 214(11), 3219–3238. [DOI:10.1084/jem.20171419] [PMID] [PMCID]
Karanfilian, L., Tosto, M. G., & Malki, K. (2020). The role of TREM2 in alzheimer's disease; evidence from transgenic mouse models. Neurobiology of Aging, 86, 39–53.[DOI:10.1016/j.neurobiolaging.2019.09.004] [PMID]
Kemppainen, N., Laine, M., Laakso, M. P., Kaasinen, V., Någren, K., & Vahlberg, T., et al. (2003). Hippocampal dopamine D2 receptors correlate with memory functions in Alzheimer's disease. The European Journal of Neuroscience, 18(1), 149–154. [DOI:10.1046/j.1460-9568.2003.02716.x] [PMID]
Kennedy, M. E., Stamford, A. W., Chen, X., Cox, K., Cumming, J. N., & Dockendorf, M. F., et al. (2016). The BACE1 inhibitor verubecestat (MK-8931) reduces CNS β-amyloid in animal models and in alzheimer's disease patients. Science Translational Medicine, 8(363), 363ra150. [DOI:10.1126/scitranslmed.aad9704] [PMID]
Khalil, H. S., Mitev, V., Vlaykova, T., Cavicchi, L., & Zhelev, N. (2015). Discovery and development of Seliciclib. How systems biology approaches can lead to better drug performance. Journal of Biotechnology, 202, 40–49. [DOI:10.1016/j.jbiotec.2015.02.032] [PMID]
Kounnas, M. Z., Danks, A. M., Cheng, S., Tyree, C., Ackerman, E., & Zhang, X., et al. (2010). Modulation of gamma-secretase reduces beta-amyloid deposition in a transgenic mouse model of alzheimer's disease. Neuron, 67(5), 769–780. [DOI:10.1016/j.neuron.2010.08.018] [PMID] [PMCID]
Kuo, L. M., Kuo, C. Y., Lin, C. Y., Hung, M. F., Shen, J. J., & Hwang, T. L. (2014). Intracellular glutathione depletion by oridonin leads to apoptosis in hepatic stellate cells. Molecules, 19(3), 3327–3344. [DOI:10.3390/molecules19033327] [PMID] [PMCID]
Lahiri, D. K., Maloney, B., Long, J. M., & Greig, N. H. (2014). Lessons from a BACE1 inhibitor trial: Off-site but not off base. Alzheimer's & Dementia, 10(5 Suppl), S411–S419. [DOI:10.1016/j.jalz.2013.11.004] [PMID] [PMCID]
Laird, F. M., Cai, H., Savonenko, A. V., Farah, M. H., He, K., & Melnikova, T., et al. (2005). BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. The Journal of Neuroscience, 25(50), 11693–11709. [DOI:10.1523/JNEUROSCI.2766-05.2005] [PMID] [PMCID]
Lamkanfi, M., & Dixit, V. M. (2017). A new lead to NLRP3 inhibition. The Journal of Experimental Medicine, 214(11), 3147–3149. [DOI:10.1084/jem.20171848] [PMID] [PMCID]
Lee, H. J., Woo, H., Lee, H. E., Jeon, H., Ryu, K. Y., & Nam, J. H., et al. (2020). The novel DYRK1A inhibitor KVN93 regulates cognitive function, amyloid-beta pathology, and neuroinflammation. Free Radical Biology & Medicine, 160, 575–595. [DOI:10.1016/j.freeradbiomed.2020.08.030] [PMID]
Liu, C. C., Liu, C. C., Kanekiyo, T., Xu, H., & Bu, G. (2013). Apolipoprotein E and alzheimer disease: Risk, mechanisms and therapy. Nature Reviews. Neurology, 9(2), 106–118.[DOI:10.1038/nrneurol.2012.263] [PMID] [PMCID]
Liu, C. C., Zhao, N., Fu, Y., Wang, N., Linares, C., & Tsai, C. W., et al. (2017). ApoE4 accelerates early seeding of amyloid pathology. Neuron, 96(5), 1024–1032.e3. [DOI:10.1016/j.neuron.2017.11.013] [PMID] [PMCID]
Liu, F., Su, Y., Li, B., Zhou, Y., Ryder, J., & Gonzalez-DeWhitt, P., et al. (2003). Regulation of amyloid precursor protein (APP) phosphorylation and processing by p35/Cdk5 and p25/Cdk5. FEBS Letters, 547(1-3), 193–196. [DOI:10.1016/s0014-5793(03)00714-2] [PMID]
Liu, M., Kou, L., Bin, Y., Wan, L., & Xiang, J. (2016). Complicated function of dopamine in Aβ-related neurotoxicity: Dual interactions with Tyr10 and SNK(26-28) of Aβ. Journal of Inorganic Biochemistry, 164, 119–128. [DOI:10.1016/j.jinorgbio.2016.09.007] [PMID]
Lorenzl, S., Albers, D. S., Relkin, N., Ngyuen, T., Hilgenberg, S. L., & Chirichigno, J., et al. (2003). Increased plasma levels of matrix metalloproteinase-9 in patients with alzheimer's disease. Neurochemistry International, 43(3), 191–196. [DOI:10.1016/s0197-0186(03)00004-4] [PMID]
Madani, R., Poirier, R., Wolfer, D. P., Welzl, H., Groscurth, P., & Lipp, H. P., et al. (2006). Lack of neprilysin suffices to generate murine amyloid-like deposits in the brain and behavioral deficit in vivo. Journal of Neuroscience Research, 84(8), 1871–1878. [DOI:10.1002/jnr.21074] [PMID]
Madav, Y., Wairkar, S., & Prabhakar, B. (2019). Recent therapeutic strategies targeting beta amyloid and tauopathies in Alzheimer's disease. Brain Research Bulletin, 146, 171–184.[DOI:10.1016/j.brainresbull.2019.01.004] [PMID]
Manakadan, A. A., Suja, S. T., Silvipriya, K. S., & Chithra, J. (2015). A Computational in silico approach for identification of indian herbs for the treatment of alzheimer’s disease. Research Journal of Pharmaceutical, Biological and Chemical Sciences, 6(1), 414-422. [Link]
Marchetti, C., Swartzwelter, B., Koenders, M. I., Azam, T., Tengesdal, I. W., & Powers, N., et al. (2018). NLRP3 inflammasome inhibitor OLT1177 suppresses joint inflammation in murine models of acute arthritis. Arthritis Research & Therapy, 20(1), 169.[DOI:10.1186/s13075-018-1664-2] [PMID] [PMCID]
Marr, R. A., & Hafez, D. M. (2014). Amyloid-beta and alzheimer’s disease: The role of neprilysin-2 in amyloid-beta clearance. Frontiers in Aging Neuroscience, 6, 187. [DOI:10.3389/fnagi.2014.00187]
Martone, R. L., Zhou, H., Atchison, K., Comery, T., Xu, J. Z., & Huang, X., et al. (2009). Begacestat (GSI-953): A novel, selective thiophene sulfonamide inhibitor of amyloid precursor protein gamma-secretase for the treatment of Alzheimer's disease. The Journal of Pharmacology and Experimental Therapeutics, 331(2), 598–608. [DOI:10.1124/jpet.109.152975] [PMID]
Martorana, A., & Koch, G. (2014). Is dopamine involved in Alzheimer’s disease? Frontiers in Aging Neuroscience, 6, 252. [DOI:10.3389/fnagi.2014.00252]
Maruyama, M., Higuchi, M., Takaki, Y., Matsuba, Y., Tanji, H., & Nemoto, M., et al. (2005). Cerebrospinal fluid neprilysin is reduced in prodromal Alzheimer's disease. Annals of Neurology, 57(6), 832–842.[DOI:10.1002/ana.20494] [PMID]
Melchior, B., Mittapalli, G. K., Lai, C., Duong-Polk, K., Stewart, J., & Güner, B., et al. (2019). Tau pathology reduction with SM07883, a novel, potent, and selective oral DYRK1A inhibitor: A potential therapeutic for alzheimer's disease. Aging Cell, 18(5), e13000.[DOI:10.1111/acel.13000] [PMID] [PMCID]
Menn, B., Bach, S., Blevins, T. L., Campbell, M., Meijer, L., & Timsit, S. (2010). Delayed treatment with systemic (S)-roscovitine provides neuroprotection and inhibits in vivo CDK5 activity increase in animal stroke models. Plos One, 5(8), e12117.[DOI:10.1371/journal.pone.0012117] [PMID] [PMCID]
Morris, J. C., Roe, C. M., Xiong, C., Fagan, A. M., Goate, A. M., & Holtzman, D. M., et al. (2010). APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Annals of Neurology, 67(1), 122–131.[DOI:10.1002/ana.21843] [PMID] [PMCID]
Namba, Y., & Ikeda, K. (1991). [Apolipoprotein B immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in senile dementia of alzheimer type (Japanese)]. Rinsho shinkeigaku = Clinical neurology, 31(8), 826–830. [PMID]
Neumann, U., Ufer, M., Jacobson, L. H., Rouzade-Dominguez, M. L., Huledal, G., & Kolly, C., et al. (2018). The BACE1 inhibitor CNP520 for prevention trials in alzheimer's disease. EMBO Molecular Medicine, 10(11), e9316. [DOI:10.15252/emmm.201809316] [PMID] [PMCID]
Nobili, A., Latagliata, E. C., Viscomi, M. T., Cavallucci, V., Cutuli, D., & Giacovazzo, G., et al. (2017). Dopamine neuronal loss contributes to memory and reward dysfunction in a model of alzheimer's disease. Nature Communications, 8, 14727. [DOI:10.1038/ncomms14727] [PMID] [PMCID]
Octave J. N. (1995). The amyloid peptide and its precursor in Alzheimer's disease. Reviews in the Neurosciences, 6(4), 287–316. [DOI:10.1515/REVNEURO.1995.6.4.287] [PMID]
Pan, X., Kaminga, A. C., Wen, S. W., Wu, X., Acheampong, K., & Liu, A. (2019). Dopamine and dopamine receptors in alzheimer's disease: A systematic review and network meta-analysis. Frontiers in Aging Neuroscience, 11, 175. [DOI:10.3389/fnagi.2019.00175] [PMID] [PMCID]
Panza, F., Frisardi, V., Imbimbo, B. P., Capurso, C., Logroscino, G., & Sancarlo, D.,et al. (2010). Review: γ-secretase inhibitors for the treatment of alzheimer's disease: The current state. Cns Neuroscience & Therapeutics, 16(5), 272–284. [DOI:10.1111/j.1755-5949.2010.00164.x] [PMID] [PMCID]
Pathak, A., Rohilla, A., Gupta, T., Akhtar, M. J., Haider, M. R., & Sharma, K., et al. (2018). DYRK1A kinase inhibition with emphasis on neurodegeneration: A comprehensive evolution story-cum-perspective. European Journal of Medicinal Chemistry, 158, 559–592. [DOI:10.1016/j.ejmech.2018.08.093] [PMID]
Pavo, N., Prausmüller, S., Bartko, P. E., Goliasch, G., & Hülsmann, M. (2020). Neprilysin as a biomarker: Challenges and opportunities. Cardiac Failure Review, 6, e23. [DOI:10.15420/cfr.2019.21] [PMID] [PMCID]
Rajendran, L., Schneider, A., Schlechtingen, G., Weidlich, S., Ries, J., Braxmeier, T., & Schwille, P., et al. (2008). Efficient inhibition of the alzheimer's disease beta-secretase by membrane targeting. Science, 320(5875), 520–523. [DOI:10.1126/science.1156609] [PMID]
Reeves, S., McLachlan, E., Bertrand, J., D'Antonio, F., Brownings, S., & Nair, A., et al. (2017). Therapeutic window of dopamine D2/3 receptor occupancy to treat psychosis in alzheimer's disease. Brain, 140(4), 1117–1127. [DOI:10.1093/brain/aww359] [PMID]
Reinhardt, L., Kordes, S., Reinhardt, P., Glatza, M., Baumann, M., & Drexler, H. C. A., et al. (2019). Dual inhibition of GSK3β and CDK5 protects the cytoskeleton of neurons from neuroinflammatory-mediated degeneration in vitro and in vivo. Stem Cell Reports, 12(3), 502–517. [DOI:10.1016/j.stemcr.2019.01.015] [PMID] [PMCID]
Rinne, J. O., Säkö, E., Paljärvi, L., Mölsä, P. K., & Rinne, U. K. (1986). Brain dopamine D-2 receptors in senile dementia. Journal of Neural Transmission, 65(1), 51–62. [DOI:10.1007/BF01249611] [PMID]
Rizos, C. V., Elisaf, M. S., Mikhailidis, D. P., & Liberopoulos, E. N. (2009). How safe is the use of thiazolidinediones in clinical practice?. Expert Opinion on Drug Safety, 8(1), 15–32.[DOI:10.1517/14740330802597821] [PMID]
Rizzi, L., Rosset, I., & Roriz-Cruz, M. (2014). Global epidemiology of dementia: Alzheimer's and vascular types. BioMed Research International, 2014, 908915. [DOI:10.1155/2014/908915] [PMID] [PMCID]
Rodrigues, R. P., & Silva, C. H. T. P. (2017). Discovery of potential neurodegenerative inhibitors in alzheimer’s disease by casein kinase 1 structure-based virtual screening. Medicinal Chemistry Research, 26(12), 3274-3285. [DOI:10.1007/s00044-017-2020-9]
Rosenberg, P. B., Nowrangi, M. A., & Lyketsos, C. G. (2015). Neuropsychiatric symptoms in Alzheimer's disease: What might be associated brain circuits?. Molecular Aspects of Medicine, 43-44, 25–37. [DOI:10.1016/j.mam.2015.05.005] [PMID] [PMCID]
Safieh, M., Korczyn, A. D., & Michaelson, D. M. (2019). ApoE4: An emerging therapeutic target for Alzheimer's disease. BMC Medicine, 17(1), 64. [DOI:10.1186/s12916-019-1299-4] [PMID] [PMCID]
Sakamoto, K., Matsuki, S., Matsuguma, K., Yoshihara, T., Uchida, N., & Azuma, F., et al. (2017). BACE1 inhibitor lanabecestat (AZD3293) in a phase 1 study of healthy japanese subjects: Pharmacokinetics and effects on plasma and cerebrospinal fluid Aβ peptides. Journal of Clinical Pharmacology, 57(11), 1460–1471. [DOI:10.1002/jcph.950] [PMID]
Schiering, N., D'Arcy, A., Villard, F., Ramage, P., Logel, C., & Cumin, F., et al. (2016). Structure of neprilysin in complex with the active metabolite of sacubitril. Scientific Reports, 6, 27909. [DOI:10.1038/srep27909] [PMID] [PMCID]
Schneider L. (2020). A resurrection of aducanumab for Alzheimer's disease. The Lancet. Neurology, 19(2), 111–112.[DOI:10.1016/S1474-4422(19)30480-6] [PMID]
Scott, J. D., Li, S. W., Brunskill, A. P., Chen, X., Cox, K., & Cumming, J. N., et al. (2016). Discovery of the 3-Imino-1,2,4-thiadiazinane 1,1-Dioxide derivative verubecestat (MK-8931)-A β-site amyloid precursor protein cleaving enzyme 1 Inhibitor for the treatment of alzheimer's disease. Journal of Medicinal Chemistry, 59(23), 10435–10450. [DOI:10.1021/acs.jmedchem.6b00307] [PMID]
Seo, J., Kritskiy, O., Watson, L. A., Barker, S. J., Dey, D., & Raja, W. K., et al. (2017). Inhibition of p25/Cdk5 attenuates tauopathy in mouse and iPSC models of frontotemporal dementia. The Journal of Neuroscience, 37(41), 9917–9924. [DOI:10.1523/JNEUROSCI.0621-17.2017] [PMID] [PMCID]
Serrano-Pozo, A., Frosch, M. P., Masliah, E., & Hyman, B. T. (2011). Neuropathological alterations in alzheimer disease. Cold Spring Harbor Perspectives in Medicine, 1(1), a006189. [DOI:10.1101/cshperspect.a006189] [PMID] [PMCID]
Sevigny, J., Chiao, P., Bussière, T., Weinreb, P. H., Williams, L., & Maier, M., et al. (2016). The antibody aducanumab reduces Aβ plaques in alzheimer's disease. Nature, 537(7618), 50–56. [DOI:10.1038/nature19323] [PMID]
Shah, K., & Lahiri, D. K. (2014). Cdk5 activity in the brain - multiple paths of regulation. Journal of Cell Science, 127(Pt 11), 2391–2400. [DOI:10.1242/jcs.147553] [PMID] [PMCID]
Shankarappa, S. A., Tsui, J. H., Kim, K. N., Reznor, G., Dohlman, J. C., & Langer, R., et al. (2012). Prolonged nerve blockade delays the onset of neuropathic pain. Proceedings of the National Academy of Sciences of the United States of America, 109(43), 17555–17560. [DOI:10.1073/pnas.1214634109] [PMID] [PMCID]
Shukla, V., Skuntz, S., & Pant, H. C. (2012). Deregulated Cdk5 activity is involved in inducing Alzheimer's disease. Archives of Medical Research, 43(8), 655–662. [DOI:10.1016/j.arcmed.2012.10.015] [PMID] [PMCID]
Song, L., Pei, L., Yao, S., Wu, Y., & Shang, Y. (2017). NLRP3 inflammasome in neurological diseases, from functions to therapies. Frontiers in Cellular Neuroscience, 11, 63. [DOI:10.3389/fncel.2017.00063]
Soto, M., Reynish, E., Nourhashémi, F., & Vellas, B. (2007). [Clinical aspects of Alzheimer disease (French)]. Presse Medicale, 36(10 Pt 2), 1491–1499. [DOI:10.1016/j.lpm.2007.04.024] [PMID]
Souchet, B., Audrain, M., Billard, J. M., Dairou, J., Fol, R., & Orefice, N. S., et al. (2019). Inhibition of DYRK1A proteolysis modifies its kinase specificity and rescues alzheimer phenotype in APP/PS1 mice. Acta Neuropathologica Communications, 7(1), 46. [DOI:10.1186/s40478-019-0678-6] [PMID] [PMCID]
Stotani, S., Giordanetto, F., & Medda, F. (2016). DYRK1A inhibition as potential treatment for alzheimer's disease. Future Medicinal Chemistry, 8(6), 681–696. [DOI:10.4155/fmc-2016-0013] [PMID]
Sundaram, S., Nagaraj, S., Mahoney, H., Portugues, A., Li, W., & Millsaps, K., et al. (2019). Inhibition of casein kinase 1δ/εimproves cognitive-affective behavior and reduces amyloid load in the APP-PS1 mouse model of alzheimer's disease. Scientific Reports, 9(1), 13743. [DOI:10.1038/s41598-019-50197-x] [PMID] [PMCID]
Tan, M. S., Yu, J. T., Jiang, T., Zhu, X. C., & Tan, L. (2013). The NLRP3 inflammasome in alzheimer's disease. Molecular Neurobiology, 48(3), 875–882. [DOI:10.1007/s12035-013-8475-x] [PMID]
Tang, W., Lv, B., Yang, B., Chen, Y., Yuan, F., & Ma, L., et al. (2019). TREM2 acts as a tumor suppressor in hepatocellular carcinoma by targeting the PI3K/Akt/β-catenin pathway. Oncogenesis, 8(2), 9. [DOI:10.1038/s41389-018-0115-x] [PMID] [PMCID]
Taniguchi, S., Fujita, Y., Hayashi, S., Kakita, A., Takahashi, H., & Murayama, S., et al. (2001). Calpain-mediated degradation of p35 to p25 in postmortem human and rat brains. FEBs Letters, 489(1), 46–50. [DOI:10.1016/s0014-5793(00)02431-5] [PMID]
Timmers, M., Streffer, J. R., Russu, A., Tominaga, Y., Shimizu, H., & Shiraishi, A., et al. (2018). Pharmacodynamics of atabecestat (JNJ-54861911), an oral BACE1 inhibitor in patients with early Alzheimer's disease: Randomized, double-blind, placebo-controlled study. Alzheimer's Research & Therapy, 10(1), 85. [DOI:10.1186/s13195-018-0415-6] [PMID] [PMCID]
Toldo, S., Mauro, A. G., Cutter, Z., Van Tassell, B. W., Mezzaroma, E., & Del Buono, M. G., et al. (2019). The NLRP3 inflammasome inhibitor, OLT1177 (Dapansutrile), reduces infarct size and preserves contractile function after ischemia reperfusion injury in the mouse. Journal of Cardiovascular Pharmacology, 73(4), 215–222. [DOI:10.1097/FJC.0000000000000658] [PMID]
Vassar, R., & Kandalepas, P. C. (2011). The β-secretase enzyme BACE1 as a therapeutic target for alzheimer's disease. Alzheimer's Research & Therapy, 3(3), 20. [DOI:10.1186/alzrt82] [PMID] [PMCID]
Velazquez, R., Meechoovet, B., Ow, A., Foley, C., Shaw, A., & Smith, B., et al. (2019). Chronic dyrk1 inhibition delays the onset of AD-like pathology in 3xTg-AD mice. Molecular Neurobiology, 56(12), 8364–8375. [DOI:10.1007/s12035-019-01684-9] [PMID]
Vodovar, N., Paquet, C., Mebazaa, A., Launay, J. M., Hugon, J., & Cohen-Solal, A. (2015). Neprilysin, cardiovascular, and Alzheimer's diseases: The therapeutic split?. European Heart Journal, 36(15), 902–905. [DOI:10.1093/eurheartj/ehv015] [PMID]
Wang, S., Mustafa, M., Yuede, C. M., Salazar, S. V., Kong, P., & Long, H., et al. (2020). Anti-human TREM2 induces microglia proliferation and reduces pathology in an alzheimer's disease model. The Journal of Experimental Medicine, 217(9), e20200785. [DOI:10.1084/jem.20200785] [PMID] [PMCID]
Wang, S., Yuan, Y. H., Chen, N. H., & Wang, H. B. (2019). The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson's disease. International Immunopharmacology, 67, 458–464. [DOI:10.1016/j.intimp.2018.12.019] [PMID]
Wang, W. Y., Tan, M. S., Yu, J. T., & Tan, L. (2015). Role of pro-inflammatory cytokines released from microglia in alzheimer's disease. Annals of Translational Medicine, 3(10), 136.[DOI:10.3978/j.issn.2305-5839.2015.03.49] [PMID] [PMCID]
Wang, X. X., Tan, M. S., Yu, J. T., & Tan, L. (2014). Matrix metalloproteinases and their multiple roles in alzheimer's disease. BioMed Research International, 2014, 908636. [DOI:10.1155/2014/908636] [PMID] [PMCID]
Wang, Y., Zhao, J., Guo, F. L., Gao, X., Xie, X., & Liu, S., et al. (2020). Metformin ameliorates synaptic defects in a mouse model of AD by inhibiting cdk5 activity. Frontiers in Cellular Neuroscience, 14, 170. [DOI:10.3389/fncel.2020.00170] [PMID] [PMCID]
Webster, C. I., Burrell, M., Olsson, L. L., Fowler, S. B., Digby, S., & Sandercock, A., et al. (2014). Engineering neprilysin activity and specificity to create a novel therapeutic for alzheimer's disease. Plos One, 9(8), e104001. [DOI:10.1371/journal.pone.0104001] [PMID] [PMCID]
Wilkaniec, A., Czapski, G. A., & Adamczyk, A. (2016). Cdk5 at crossroads of protein oligomerization in neurodegenerative diseases: Facts and hypotheses. Journal of Neurochemistry, 136(2), 222–233. [DOI:10.1111/jnc.13365] [PMID]
Wilkaniec, A., Gąssowska-Dobrowolska, M., Strawski, M., Adamczyk, A., & Czapski, G. A. (2018). Inhibition of cyclin-dependent kinase 5 affects early neuroinflammatory signalling in murine model of amyloid beta toxicity. Journal of Neuroinflammation, 15(1), 1. [DOI:10.1186/s12974-017-1027-y] [PMID] [PMCID]
Wolfe M. S. (2012). γ-Secretase inhibitors and modulators for alzheimer's disease. Journal of Neurochemistry, 120(Suppl 1), 89–98. [DOI:10.1111/j.1471-4159.2011.07501.x] [PMID] [PMCID]
Wolfe M. S. (2014). Unlocking truths of γ-secretase in Alzheimer's disease: What is the translational potential?. Future Neurology, 9(4), 419–429. [DOI:10.2217/fnl.14.35] [PMID] [PMCID]
Yamazaki, Y., Zhao, N., Caulfield, T. R., Liu, C. C., & Bu, G. (2019). Apolipoprotein E and alzheimer disease: Pathobiology and targeting strategies. Nature Reviews. Neurology, 15(9), 501–518. [DOI:10.1038/s41582-019-0228-7] [PMID] [PMCID]
Yan, R., Bienkowski, M. J., Shuck, M. E., Miao, H., Tory, M. C., & Pauley, A. M., et al. (1999). Membrane-anchored aspartyl protease with alzheimer's disease beta-secretase activity. Nature, 402(6761), 533–537 [DOI:10.1038/990107] [PMID]
Yang, E. J., Ahn, Y. S., & Chung, K. C. (2001). Protein kinase Dyrk1 activates cAMP response element-binding protein during neuronal differentiation in hippocampal progenitor cells. The Journal of Biological Chemistry, 276(43), 39819–39824. [DOI:10.1074/jbc.M104091200] [PMID]
Yang, S. J., Shao, G. F., Chen, J. L., & Gong, J. (2018). The NLRP3 inflammasome: An important driver of neuroinflammation in hemorrhagic stroke. Cellular and Molecular Neurobiology, 38(3), 595–603. [DOI:10.1007/s10571-017-0526-9] [PMID]
Yang, Y., Wang, H., Kouadir, M., Song, H., & Shi, F. (2019). Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death & Disease, 10(2), 128.[DOI:10.1038/s41419-019-1413-8] [PMID] [PMCID]
Yasojima, K., Kuret, J., DeMaggio, A. J., McGeer, E., & McGeer, P. L. (2000). Casein kinase 1 delta mRNA is upregulated in alzheimer disease brain. Brain Research, 865(1), 116–120. [DOI:10.1016/s0006-8993(00)02200-9] [PMID]
Yildiz-Unal, A., Korulu, S., & Karabay, A. (2015). Neuroprotective strategies against calpain-mediated neurodegeneration. Neuropsychiatric Disease and Treatment, 11, 297–310. [DOI:10.2147/NDT.S78226] [PMID] [PMCID]
Yin, J., Zhao, F., Chojnacki, J. E., Fulp, J., Klein, W. L., & Zhang, S., et al. (2018). NLRP3 inflammasome inhibitor ameliorates amyloid pathology in a mouse model of alzheimer's disease. Molecular Neurobiology, 55(3), 1977–1987.[DOI:10.1007/s12035-017-0467-9] [PMID] [PMCID]
Yin, K. J., Cirrito, J. R., Yan, P., Hu, X., Xiao, Q., & Pan, X., et al. (2006). Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. The Journal of Neuroscience, 26(43), 10939–10948. [DOI:10.1523/JNEUROSCI.2085-06.2006] [PMID] [PMCID]
Yong, V. W., Agrawal, S. M., & Stirling, D. P. (2007). Targeting MMPs in acute and chronic neurological conditions. Neurotherapeutic, 4(4), 580–589. [DOI:10.1016/j.nurt.2007.07.005] [PMID] [PMCID]
Yoshiyama, Y., Asahina, M., & Hattori, T. (2000). Selective distribution of matrix metalloproteinase-3 (MMP-3) in alzheimer's disease brain. Acta Neuropathologica, 99(2), 91–95. [DOI:10.1007/pl00007428] [PMID]
Zahid, A., Li, B., Kombe, A. J. K., Jin, T., & Tao, J. (2019). Pharmacological inhibitors of the NLRP3 inflammasome. Frontiers in Immunology, 10, 2538. [DOI:10.3389/fimmu.2019.02538] [PMID] [PMCID]
Zhang, Y. W., Thompson, R., Zhang, H., & Xu, H. (2011). APP processing in alzheimer's disease. Molecular Brain, 4, 3. [DOI:10.1186/1756-6606-4-3] [PMID] [PMCID]
Zheng, H., Cheng, B., Li, Y., Li, X., Chen, X., & Zhang, Y. W. (2018). TREM2 in alzheimer's disease: Microglial survival and energy metabolism. Frontiers in Aging Neuroscience, 10, 395. [DOI:10.3389/fnagi.2018.00395] [PMID] [PMCID]
Zwan, M. D., Bouwman, F. H., Konijnenberg, E., van der Flier, W. M., Lammertsma, A. A., & Verhey, F. R., et al. (2017). Diagnostic impact of [18F]flutemetamol PET in early-onset dementia. Alzheimer's Research & Therapy, 9(1), 2. [DOI:10.1186/s13195-016-0228-4] [PMID] [PMCID]
Type of Study: Review |
Subject:
Cellular and molecular Neuroscience
Received: 2021/04/29 | Accepted: 2021/06/6 | Published: 2024/01/1
Received: 2021/04/29 | Accepted: 2021/06/6 | Published: 2024/01/1
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
