

Volume 12, Issue 4 (July & August 2021)
BCN 2021, 12(4): 563-568 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Noorian S, Hamzehlou S, Rabbani A, Sotoudeh A, Pour Rostami K, Savad S. The Role of Thyroid Function Tests in Diagnosing Allan-herndon-dudley Syndrome Revisited: A Novel Iran-based Mutation. BCN 2021; 12 (4) :563-568
URL: http://bcn.iums.ac.ir/article-1-1552-en.html
URL: http://bcn.iums.ac.ir/article-1-1552-en.html
Shahab Noorian1 

 , Sepideh Hamzehlou *2 , Ali Rabbani3 , Arya Sotoudeh3 , Kioumars Pour Rostami1 , Shahram Savad2
, Sepideh Hamzehlou *2 , Ali Rabbani3 , Arya Sotoudeh3 , Kioumars Pour Rostami1 , Shahram Savad2
, Sepideh Hamzehlou *2 , Ali Rabbani3 , Arya Sotoudeh3 , Kioumars Pour Rostami1 , Shahram Savad2
1- Department of Pediatric Endocrinology and Metabolism, Bahonar Hospital, Alborz University of Medical Sciences, Karaj, Iran.
2- Department of Medical Genetics, School of Medicine, Tehran University of Medical Sciences, Tehran, Iran.
3- Growth and Development Research Center, Children’s Medical Center, Tehran University of Medical Sciences, Tehran, Iran.
2- Department of Medical Genetics, School of Medicine, Tehran University of Medical Sciences, Tehran, Iran.
3- Growth and Development Research Center, Children’s Medical Center, Tehran University of Medical Sciences, Tehran, Iran.
Full-Text [PDF 701 kb]
| Abstract (HTML)
Full-Text:
1. Introduction
Allan-Herndon-Dudley Syndrome (AHDS) is an X-linked recessive intellectual disability condition, characterized by muscular hypoplasia, hypotonia, spastic paraplegia (Dumitrescu, Liao, Best, Brockmann, & Refetoff, 2004). AHDS is a rare disease with an overall prevalence of less than one in a million. The disease manifestations appear in the neonatal period or early infancy.
AHDS is caused by mutations in the SLC16A2 gene (Xq13.2). This gene, expressed in the brain, encodes for Monocarboxylate Transporter 8 (MCT8) and is a specific transporter of thyroid hormone T3 into nerve cells (Schwartz et al., 2005). Gene mutations altering the structure and function of the SLC16A2 protein effectively disrupt T3 transport into nerve cells; thus, it disrupts normal brain development, resulting in intellectual disability and problems with movement. Moreover, T3 is not taken up by nerve cells; therefore, excess amounts of this hormone continue to circulate in the bloodstream and is a hallmark of AHDS diagnosis.
Diagnosis is based on clinical manifestations, delayed myelination of the brain on MRI radiological examination, and the presence of altered thyroid function tests. Affected males have abnormally high T3 levels, low to normal T4 levels, and normal to slightly elevated TSH levels. Diagnosis is confirmed by molecular genetic testing revealing mutations in the SLC16A2 gene. :as char:acteristics of X-linked recessive inheritance, the female can pass on the mutated gene; however, it usually does not experience signs and symptoms of the disorder. The carriers of SLC16A2 mutations have normal thyroid function tests and healthy intelligence and do not experience movement problems. In this study, we reported a 3.5-year-old boy with AHDS diagnosis and a novel synonymous missense mutation in the SLC16A2 gene manifesting normal levels of T3, T4, and TSH.
2. Case Presentation
Medical examination and genetic counseling: A 3.5-year-old boy was referred to our clinic for evaluating floppiness. He was also under the observation of a pediatric neurologist because of cerebral palsy. He was born from unrelated parents (Figure 1); delivery occurred in 38 weeks, with a birth weight of 2.9 kg and length of 50cm.
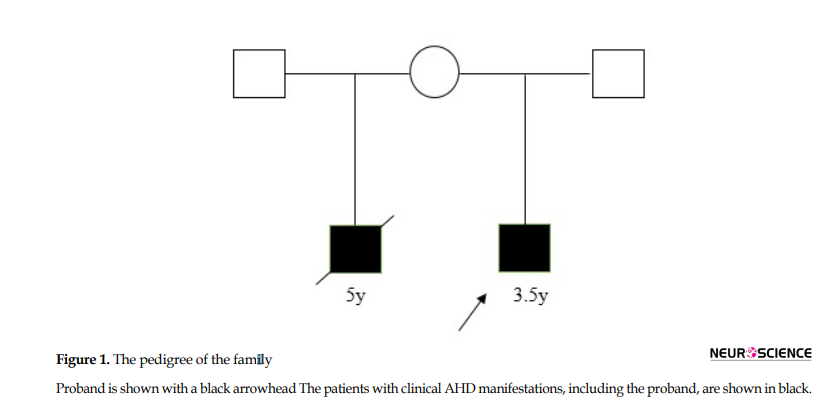
His progressive weakness was discovered a few months after birth. On our examination, he manifested a failure to thrive; his weight was 11 kg (< the 5th percentile of corresponding age) and his length was 87cm (<the 5th percentile of corresponding age). The head circumference was 47.5 cm (equal to the 50th percentile). He manifested excessive drooling, obvious head lag, and global developmental delay implying serious neurological damage. He could follow objects and pay attention to sounds but was unable to walk, speak, and feed independently. Other neuromuscular findings involved generalized hypotonia, the hypotonia of limbs, and increased deep tendon reflexes. Genitalia was prepubertal and testes were descended and palpable. Laboratory tests revealed normal metabolic and lipid profile, normal liver function tests, and normal levels of free triiodothyronine (T3) (3.87 pg/mL within the normal range of 3.7-4.4 pg/mL), free thyroxine (T4) (1.23 ng/dL within the normal range of 1.1-1.4 ng/dL) and TSH (2.34 mIU/L within the normal range of 1.75-3.5 mIU/L). Auditory Brainstem Response (ABR) test data revealed no abnormal latency time; however, brain MRI manifested remarkable delayed myelination of the brain white matter. The mother disclosed that from another marriage, she had another son with similar phenotypes who had died at the age of 5 years.
Whole exome sequencing: This familial occurrence suggested an X-linked recessive disorder; however, because AHDS was not suspected before analysis, exome sequencing was performed. After obtaining our Institutional Ethical Committee’s approval and receiving written informed consent from the patient’s family, the patient’s blood sample was obtained and genomic DNA was extracted. Clinical exome sequencing was performed using the Illumina HiSeq4000 sequencing panel.
3. Results
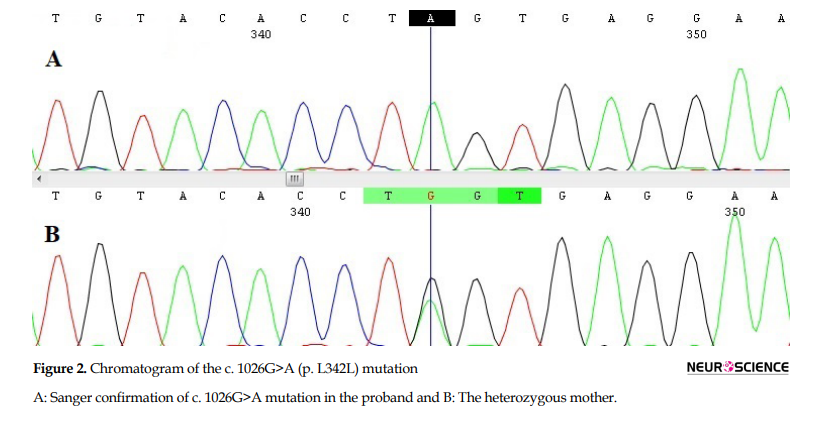
A novel single nucleotide alteration in exon 3 (c.1026G>A) was considered as the mutation related to the patient’s condition. The mutation does not cause any changes in the amino acid sequence (p. L342L). This variant is not reported in the 1000 genomes database and ExAC browser. Using Mutation Taster, the variant was predicted as damaging. Using HSF (Human Splicing Finder), the mutation was predicted to alter a wild-type splicing donor site, most probably affecting splicing through the alteration of an Exonic Splicing Enhancer (ESE). This mutation was confirmed in the patient by Sanger sequencing (Figure 2A). This mutation was verified in the patient’s mother (Figure 2B). Therefore, the diagnosis of AHDS was confirmed in the family.
4. Discussion
The AHDS is a rare X-linked recessive intellectual disability condition. The diagnosis is primarily based on clinical findings and altered thyroid hormone levels. Although the increased T3 levels are mentioned as an obligate finding in some studies (Friesema, Visser, Visser, & Endocrinology, 2010; Langley, Trau, Bean, Narravula, & Schrier Vergano, 2015), to the best of our knowledge, 3 studies are supporting the normal levels of thyroid hormones in AHDS patients (Boccone, Dessì, Meloni, & Loudianos, 2013; Shimojima et al., 2016; Tsurusaki et al., 2011). In this study, we reported a 3.5-year-old boy manifesting AHDS and normal thyroid function tests with a missense mutation in the SLC16A2 gene affecting splicing. In contrast to our finding, other studies reported different non-synonymous variants, one in exon 3 similar to our finding.
The reason why some patients with AHDS manifest normal thyroid hormones remains unclear. A previous study indicated a single mutation in the SLC16A2 gene in two males in a family caused phenotypic variability, as follows: the infant male showed increased T3 levels and the adult male presented normal T3 levels (Boccone et al., 2013). Boccone et al. discussed that this variable expressivity could probably be attributed to the age of the patients. From a genetic point of view, variable expressivity refers to the range of signs and symptoms that can occur in different individuals with the same genetic condition, i.e., the same pathogenic mutation in the SLC16A2 gene. Variable expressivity is probably caused by a combination of genetic (interactions with other loci), age, environmental, and lifestyle factors, most of which remained unidentified. Variable expressivity of a genetic disease makes it challenging to diagnose.
Clinical considerations: The normal thyroid hormones profile in some AHDS patients implies clinicians should consider AHDS as a candidate diagnosis in patients with X-linked recessive intellectual disability syndrome with neuromuscular involvements irrespective of the levels of thyroid hormones. Screening for mutations in the SLC16A2 gene in these cases obviates the need for expensive Whole Exome Sequencing (WES). This is particularly important in developing countries where WES is costly due to limited resources. Furthermore, this screening is essential from the view of genetic counseling. Since there is no treatment available for this disease and the disease compromises the quality of life by affecting the ability of the patient to live independently; genetic counseling to prevent the birth of an affected child is of significance. Affected families should be informed that prenatal or preimplantation diagnosis of a male with AHDS is possible if the mutation in his mother is identified. In our case, the ultimate diagnosis of AHDS in one infant uncovered his previously undiagnosed affected brother who had expired of the disease. If the diagnosis of AHDS was considered in the first brother, it could have led to prenatal or preimplantation diagnosis to prevent the birth of a second affected child.
5. Conclusion
In brief, altered levels of thyroid hormones are a notable but not necessary marker for the diagnosis of AHDS. Due to phenotypic variations in the levels of thyroid hormones in AHDS patients, the disorder is often under-diagnosed imposing economic and mental burden. Therefore, the candidate diagnosis of AHDS should be considered in patients with X-linked recessive intellectual disability syndrome with neuromuscular involvements irrespective of levels of thyroid hormones.
Ethical Considerations
Compliance with ethical guidelines
All ethical principles are considered in this article. The participants were informed of the purpose of the research and its implementation stages. They were also assured about the confidentiality of their information. They were free to leave the study whenever they wished, and if desired, the research results would be available to them. Written consent has been obtained from the subjects. Principles of the Helsinki Convention were also observed.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
All authors equally contributed to preparing this article.
Conflict of interest
The authors declared no conflict of interest.
References
Boccone, L., Dessì, V., Meloni, A., & Loudianos, G. (2013). Allan-Herndon-Dudley Syndrome (AHDS) in two consecutive generations caused by a missense MCT8 gene mutation. Phenotypic variability with the presence of normal serum T3 levels. European Journal of Medical Genetics, 56(4), 207-10. [DOI:10.1016/j.ejmg.2013.02.001] [PMID]
Dumitrescu, A. M., Liao, X. H., Best, T. B., Brockmann, K., & Refetoff, S. (2004). A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. American Journal of Human Genetics, 74(1), 168-75. [DOI:10.1086/380999] [PMID] [PMCID]
Friesema, E. C. H., Visser, W. E., & Visser, T. J. (2010). Genetics and phenomics of thyroid hormone transport by MCT8. Molecular and Cellular Endocrinology, 322(1-2), 107-13. [DOI:10.1016/j.mce.2010.01.016] [PMID]
Langley, K. G., Trau, S., Bean, L. J. H., Narravula, A., & Schrier Vergano, S. A. (2015). A 7‐month‐old male with Allan‐Herndon‐Dudley syndrome and the power of T3. American Journal of Medical Genetics, 167(5), 1117-20. [DOI:10.1002/ajmg.a.36970] [PMID]
Schwartz, C. E., May, M. M., Carpenter, N. J., Rogers, R. C., Martin, J., & Bialer, M. G., et al. (2005). Allan-Herndon-Dudley syndrome and the Monocarboxylate Transporter 8 (MCT8) gene. American Journal of Human Genetics, 77(1), 41-53. [DOI:10.1086/431313] [PMID] [PMCID]
Shimojima, K., Maruyama, K., Kikuchi, M., Imai, A., Inoue, K., & Yamamoto, T. (2016). Novel SLC16A2 mutations in patients with Allan-Herndon-Dudley syndrome. Intractable & Rare Diseases Research, 5(3), 214-7. [DOI:10.5582/irdr.2016.01051] [PMID] [PMCID]
Tsurusaki, Y., Osaka, H., Hamanoue, H., Shimbo, H., Tsuji, M., & Doi, H., et al. (2011). Rapid detection of a mutation causing X-linked leucoencephalopathy by exome sequencing. Journal of Medical Genetics, 48(9), 606-9. [DOI:10.1136/jmg.2010.083535] [PMID]
Allan-Herndon-Dudley Syndrome (AHDS) is an X-linked recessive intellectual disability condition, characterized by muscular hypoplasia, hypotonia, spastic paraplegia (Dumitrescu, Liao, Best, Brockmann, & Refetoff, 2004). AHDS is a rare disease with an overall prevalence of less than one in a million. The disease manifestations appear in the neonatal period or early infancy.
AHDS is caused by mutations in the SLC16A2 gene (Xq13.2). This gene, expressed in the brain, encodes for Monocarboxylate Transporter 8 (MCT8) and is a specific transporter of thyroid hormone T3 into nerve cells (Schwartz et al., 2005). Gene mutations altering the structure and function of the SLC16A2 protein effectively disrupt T3 transport into nerve cells; thus, it disrupts normal brain development, resulting in intellectual disability and problems with movement. Moreover, T3 is not taken up by nerve cells; therefore, excess amounts of this hormone continue to circulate in the bloodstream and is a hallmark of AHDS diagnosis.
Diagnosis is based on clinical manifestations, delayed myelination of the brain on MRI radiological examination, and the presence of altered thyroid function tests. Affected males have abnormally high T3 levels, low to normal T4 levels, and normal to slightly elevated TSH levels. Diagnosis is confirmed by molecular genetic testing revealing mutations in the SLC16A2 gene. :as char:acteristics of X-linked recessive inheritance, the female can pass on the mutated gene; however, it usually does not experience signs and symptoms of the disorder. The carriers of SLC16A2 mutations have normal thyroid function tests and healthy intelligence and do not experience movement problems. In this study, we reported a 3.5-year-old boy with AHDS diagnosis and a novel synonymous missense mutation in the SLC16A2 gene manifesting normal levels of T3, T4, and TSH.
2. Case Presentation
Medical examination and genetic counseling: A 3.5-year-old boy was referred to our clinic for evaluating floppiness. He was also under the observation of a pediatric neurologist because of cerebral palsy. He was born from unrelated parents (Figure 1); delivery occurred in 38 weeks, with a birth weight of 2.9 kg and length of 50cm.
His progressive weakness was discovered a few months after birth. On our examination, he manifested a failure to thrive; his weight was 11 kg (< the 5th percentile of corresponding age) and his length was 87cm (<the 5th percentile of corresponding age). The head circumference was 47.5 cm (equal to the 50th percentile). He manifested excessive drooling, obvious head lag, and global developmental delay implying serious neurological damage. He could follow objects and pay attention to sounds but was unable to walk, speak, and feed independently. Other neuromuscular findings involved generalized hypotonia, the hypotonia of limbs, and increased deep tendon reflexes. Genitalia was prepubertal and testes were descended and palpable. Laboratory tests revealed normal metabolic and lipid profile, normal liver function tests, and normal levels of free triiodothyronine (T3) (3.87 pg/mL within the normal range of 3.7-4.4 pg/mL), free thyroxine (T4) (1.23 ng/dL within the normal range of 1.1-1.4 ng/dL) and TSH (2.34 mIU/L within the normal range of 1.75-3.5 mIU/L). Auditory Brainstem Response (ABR) test data revealed no abnormal latency time; however, brain MRI manifested remarkable delayed myelination of the brain white matter. The mother disclosed that from another marriage, she had another son with similar phenotypes who had died at the age of 5 years.
Whole exome sequencing: This familial occurrence suggested an X-linked recessive disorder; however, because AHDS was not suspected before analysis, exome sequencing was performed. After obtaining our Institutional Ethical Committee’s approval and receiving written informed consent from the patient’s family, the patient’s blood sample was obtained and genomic DNA was extracted. Clinical exome sequencing was performed using the Illumina HiSeq4000 sequencing panel.
3. Results
A novel single nucleotide alteration in exon 3 (c.1026G>A) was considered as the mutation related to the patient’s condition. The mutation does not cause any changes in the amino acid sequence (p. L342L). This variant is not reported in the 1000 genomes database and ExAC browser. Using Mutation Taster, the variant was predicted as damaging. Using HSF (Human Splicing Finder), the mutation was predicted to alter a wild-type splicing donor site, most probably affecting splicing through the alteration of an Exonic Splicing Enhancer (ESE). This mutation was confirmed in the patient by Sanger sequencing (Figure 2A). This mutation was verified in the patient’s mother (Figure 2B). Therefore, the diagnosis of AHDS was confirmed in the family.
4. Discussion
The AHDS is a rare X-linked recessive intellectual disability condition. The diagnosis is primarily based on clinical findings and altered thyroid hormone levels. Although the increased T3 levels are mentioned as an obligate finding in some studies (Friesema, Visser, Visser, & Endocrinology, 2010; Langley, Trau, Bean, Narravula, & Schrier Vergano, 2015), to the best of our knowledge, 3 studies are supporting the normal levels of thyroid hormones in AHDS patients (Boccone, Dessì, Meloni, & Loudianos, 2013; Shimojima et al., 2016; Tsurusaki et al., 2011). In this study, we reported a 3.5-year-old boy manifesting AHDS and normal thyroid function tests with a missense mutation in the SLC16A2 gene affecting splicing. In contrast to our finding, other studies reported different non-synonymous variants, one in exon 3 similar to our finding.
The reason why some patients with AHDS manifest normal thyroid hormones remains unclear. A previous study indicated a single mutation in the SLC16A2 gene in two males in a family caused phenotypic variability, as follows: the infant male showed increased T3 levels and the adult male presented normal T3 levels (Boccone et al., 2013). Boccone et al. discussed that this variable expressivity could probably be attributed to the age of the patients. From a genetic point of view, variable expressivity refers to the range of signs and symptoms that can occur in different individuals with the same genetic condition, i.e., the same pathogenic mutation in the SLC16A2 gene. Variable expressivity is probably caused by a combination of genetic (interactions with other loci), age, environmental, and lifestyle factors, most of which remained unidentified. Variable expressivity of a genetic disease makes it challenging to diagnose.
Clinical considerations: The normal thyroid hormones profile in some AHDS patients implies clinicians should consider AHDS as a candidate diagnosis in patients with X-linked recessive intellectual disability syndrome with neuromuscular involvements irrespective of the levels of thyroid hormones. Screening for mutations in the SLC16A2 gene in these cases obviates the need for expensive Whole Exome Sequencing (WES). This is particularly important in developing countries where WES is costly due to limited resources. Furthermore, this screening is essential from the view of genetic counseling. Since there is no treatment available for this disease and the disease compromises the quality of life by affecting the ability of the patient to live independently; genetic counseling to prevent the birth of an affected child is of significance. Affected families should be informed that prenatal or preimplantation diagnosis of a male with AHDS is possible if the mutation in his mother is identified. In our case, the ultimate diagnosis of AHDS in one infant uncovered his previously undiagnosed affected brother who had expired of the disease. If the diagnosis of AHDS was considered in the first brother, it could have led to prenatal or preimplantation diagnosis to prevent the birth of a second affected child.
5. Conclusion
In brief, altered levels of thyroid hormones are a notable but not necessary marker for the diagnosis of AHDS. Due to phenotypic variations in the levels of thyroid hormones in AHDS patients, the disorder is often under-diagnosed imposing economic and mental burden. Therefore, the candidate diagnosis of AHDS should be considered in patients with X-linked recessive intellectual disability syndrome with neuromuscular involvements irrespective of levels of thyroid hormones.
Ethical Considerations
Compliance with ethical guidelines
All ethical principles are considered in this article. The participants were informed of the purpose of the research and its implementation stages. They were also assured about the confidentiality of their information. They were free to leave the study whenever they wished, and if desired, the research results would be available to them. Written consent has been obtained from the subjects. Principles of the Helsinki Convention were also observed.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
All authors equally contributed to preparing this article.
Conflict of interest
The authors declared no conflict of interest.
References
Boccone, L., Dessì, V., Meloni, A., & Loudianos, G. (2013). Allan-Herndon-Dudley Syndrome (AHDS) in two consecutive generations caused by a missense MCT8 gene mutation. Phenotypic variability with the presence of normal serum T3 levels. European Journal of Medical Genetics, 56(4), 207-10. [DOI:10.1016/j.ejmg.2013.02.001] [PMID]
Dumitrescu, A. M., Liao, X. H., Best, T. B., Brockmann, K., & Refetoff, S. (2004). A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. American Journal of Human Genetics, 74(1), 168-75. [DOI:10.1086/380999] [PMID] [PMCID]
Friesema, E. C. H., Visser, W. E., & Visser, T. J. (2010). Genetics and phenomics of thyroid hormone transport by MCT8. Molecular and Cellular Endocrinology, 322(1-2), 107-13. [DOI:10.1016/j.mce.2010.01.016] [PMID]
Langley, K. G., Trau, S., Bean, L. J. H., Narravula, A., & Schrier Vergano, S. A. (2015). A 7‐month‐old male with Allan‐Herndon‐Dudley syndrome and the power of T3. American Journal of Medical Genetics, 167(5), 1117-20. [DOI:10.1002/ajmg.a.36970] [PMID]
Schwartz, C. E., May, M. M., Carpenter, N. J., Rogers, R. C., Martin, J., & Bialer, M. G., et al. (2005). Allan-Herndon-Dudley syndrome and the Monocarboxylate Transporter 8 (MCT8) gene. American Journal of Human Genetics, 77(1), 41-53. [DOI:10.1086/431313] [PMID] [PMCID]
Shimojima, K., Maruyama, K., Kikuchi, M., Imai, A., Inoue, K., & Yamamoto, T. (2016). Novel SLC16A2 mutations in patients with Allan-Herndon-Dudley syndrome. Intractable & Rare Diseases Research, 5(3), 214-7. [DOI:10.5582/irdr.2016.01051] [PMID] [PMCID]
Tsurusaki, Y., Osaka, H., Hamanoue, H., Shimbo, H., Tsuji, M., & Doi, H., et al. (2011). Rapid detection of a mutation causing X-linked leucoencephalopathy by exome sequencing. Journal of Medical Genetics, 48(9), 606-9. [DOI:10.1136/jmg.2010.083535] [PMID]
Type of Study: News and Reports |
Subject:
Cellular and molecular Neuroscience
Received: 2019/07/8 | Accepted: 2020/06/30 | Published: 2021/07/1
Received: 2019/07/8 | Accepted: 2020/06/30 | Published: 2021/07/1
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
