




BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
URL: http://bcn.iums.ac.ir/article-1-1435-en.html



2- Department of Physiology, Faculty of Medical Sciences, Tarbiat Modares University, Tehran, Iran
• Epilepsy is one of the most prevalent chronic brain disorders worldwide;
• Selective locked of A1 receptors by administration of 8-cyclopenthyle dimethylexanthine significantly reduced LFS effectiveness in recovering these parameters.
• Patients diagnosed with refractory epilepsy may qualify for surgical resection of epileptic brain tissue.
• Recently, deep brain stimulation (DBS) is used in the treatment of refractory epilepsy.
• Low-frequency electrical stimulation (LFS) is an effective pattern of DBS in both clinical and experimental studies.
Plain Language Summary
Epilepsy is one of the most prevalent chronic brain disorders worldwide characterized by the presence of recurrent, spontaneous seizures;however, seizures do not respond well or are resistant to them all.
1. Introduction
Epilepsy is one of the most prevalent chronic brain disorders worldwide, characterized by the presence of recurrent, spontaneous seizures, resulting from the uncontrolled, synchronous excitation of a neuronal population within the brain. Despite numerous available antiepileptic drugs, seizures do not respond well or are resistant to them in more than 20%-40% of the patients (French, 2007). Patients diagnosed with refractory epilepsy may qualify for surgical resection of epileptic brain tissue. However, there are serious postoperative complications following neurosurgical resection, such as surgical mortality, permanent or temporary language and motor or psychologic deficits, and possibly, lack of amelioration or worsening in seizure activity (especially in patients with tumor-associated epilepsy) (Kwan & Brodie, 2000; Lin et al., 2011).
Furthermore, over 40% of the patients with refractory epilepsy do not meet the appropriateness criteria for the surgical treatment (Kwan & Brodie, 2000). Among the epilepsy syndromes, mesial temporal lobe epilepsy is the most frequent type of focal epilepsy with intractable seizures in adulthood. In this morbidity, seizures arise from different areas of the temporal lobe, such as the hippocampus, parahippocampal gyrus, and amygdala (Engel, 2001), and pathologically are characterized by specific morphological alterations in the hippocampus (Hinterkeuser et al., 2000; Temkin, 2009).
In recent years, there has been a growing interest in application of Deep Brain Stimulation (DBS) in the treatment of refractory epilepsy. This therapeutic method involves the implantation of one or more electrodes into specific brain regions to deliver controlled electrical pulses to a target in brain to modulate neuronal activity. Although DBS is a recognized therapeutic tool that has received the U.S. Food and Drug Administration (FDA) approval for the treatment of refractory epilepsy; however, the antiepileptic mechanisms involved in its effectiveness are still largely unknown.
Low-Frequency electrical Stimulation (LFS) is an effective pattern of DBS, which has shown with anticonvulsive activity in both clinical (Kinoshita et al., 2004; Yamamoto et al., 2002; Yamamoto et al., 2006) and experimental (Gaito & Gaito, 1981; Gharib, Sayyahi, Komaki, Barkley, Sarihi, & Mirnajafi-Zadeh, 2018; Goodman, Berger, & Tcheng, 2005; Mohammad-Zadeh et al., 2007; Shahpari, Mirnajafi-Zadeh, Firoozabadi, , & Yadollahpour, 2012) studies. We have previously demonstrated that the application of LFS at a frequency of 1 Hz retards the onset or development of seizures in both in vivo and in vitro models of seizures (Ahmadirad et al., 2019; Ghasemi et al., 2019; Ghorbani, Mohammad-Zadeh, Mirnajafi-Zadeh, & Fathollahi, 2007; Jahanshahi Mirnajafi-Zadeh, Javan, Mohammad-Zadeh, & Rohani, 2009; Shahpari et al., 2012). This LFS paradigm was also able to improve learning and memory deficits following kindling acquisition (Esmaeilpour, Sheibani, Shabani, & Mirnajafi-Zadeh, 2017). Conventionally, LFS is a well-known research tool for the elimination of long-term potentiation, and induction of long-term depression of synaptic transmission. It has been proposed that LFS employs similar mechanisms to exert its inhibitory effect on seizure activity, such as endogenous adenosine system.
Adenosine, as an endogenous purine ribonucleoside, is a neuromodulator and a homeostatic regulator in the nervous system that mostly suppresses excitatory synaptic transmission and reduces neuronal excitability (de Mendonça & Ribeiro, 1997; Ribeiro, 1995). Adenosine is released during seizures and it is believed to have an important role in terminating the seizures (Dunwiddie , Hoffer, & Fredholm, 1981; During & Spencer, 1992; Ribeiro, Sebastiao, & de Mendonca, 2003; Zhou et al., 2018). Anticonvulsant activity of the adenosine system has been demonstrated in various animal models of epilepsy, and its dysfunction can cause a seizure (Alasvand Zarasvand, Mirnajafi-Zadeh, Fathollahi, & Palizvan, 2001; Boison, 2005, 2013; Chwalczuk, Rubaj, Swiader, & Czuczwar, 2008; Ekonomou, Angelatou, Vergnes, & Kostopoulos, 1998; Gasior, Borowicz, Kleinrok, & Czuczwar, 1996; Malhotra & Gupta, 1997; Siebel et al., 2015; Young & Dragunow, 1994). Antiepileptic effect of adenosine is mainly accomplished through the activation of its A1 receptors, which is the most abundant adenosine receptor in brain areas critical for epileptogenesis, such as the hippocampus (Fedele, Li, Lan, Fredholm, & Boison, 2006; Fredholm et al., 2001; Fredholm, Chen, Masino, & Vaugeois, 2005; Hosseinmardi, Mirnajafi-Zadeh, Fathollahi, & Shahabi, 2007; Zeraati, Mirnajafi-Zadeh, Fathollahi, Namvar, & Rezvani, 2006; Zhou et al., 2018).
Our previous studies have shown that the anticonvulsant effect of LFS is partly mediated through adenosine A1 receptors (Jahanshahi et al., 2009; Mohammad-Zadeh et al., 2009). In addition, LFS employs adenosine A1 receptors to inhibit seizure-induced potentiation of Field Excitatory Postsynaptic Potentials (fEPSP) in dentate gyrus of the hippocampal formation Mohammad-Zadeh et al., 2009).
In another study, we reported that the application of LFS can prevent seizure-induced hyperexcitability of CA1 pyramidal neurons in hippocampus (Ghotbedin, Janahmadi, Mirnajafi-Zadeh, Behzadi, & Semnanian, 2013). Considering the important role of adenosine A1 receptors in the anti-epileptic effect of LFS, in this study, we investigated the role of these receptors in the effectiveness of LFS applied following kindling acquisition on seizure-induced hyperexcitability of hippocampal CA1 pyramidal neurons in rats. Accordingly, we employed a semi-rapid hippocampal kindling model to induce and develop the seizures and used the whole-cell patch-clamp recording to examine electrophysiological properties of the hippocampal CA1 pyramidal neurons.
2. Methods
2.1. Animals
Male Wistar rats (45 days old) were kept in normal conditions at 23±2°C temperature and a 12:12 light:dark schedule (lights on at 7:00 A.M) with ad libitum access to food and water. Adequate measures were taken to minimize pain or discomfort. All experiments were carried out based on the ethical guidelines set by the Ethical Committee of Faculty of Medical Sciences, Tarbiat Modares University and were entirety complied with the “NIH Guide for the Care and Use of Laboratory Animals”.
2.2. Animal surgery
Animals were anesthetized with a ketamine/xylazine mixture (100/10 mg/kg, i.p.) and underwent stereotaxic implantation of a bipolar stimulating electrode in right Schaffer Collateral pathway (coordinates: A, 3.1 mm; L, 3.1 mm from Bregma and 2.8 mm below the dura), a monopolar recording electrode in CA1 area (coordinates: A, 2.8 mm; L, 1.8 mm from Bregma and 2.4 mm below the dura) of the hippocampus and a 23-gauge guide cannula was also located in the left lateral ventricle (coordinates: A, −0.9 mm; L, −1.5 mm from Bregma and 3.5 mm below the dura) (Paxinos & Watson, 2009). The exact position of the recording and stimulating electrodes was set to record a maximum slope of field EPSP in the stratum radiatum layer of the hippocampal CA1 area following the Schaffer Collateral pathway stimulation and also to confirm the correct electrodes placement. The electrodes composed of twisted stainless steel, Teflon coated wires with a diameter of 127 µm insulated, except for their tips (A-M Systems, Inc., WA, U.S.A.). Each reference and ground electrode consisted of a stainless-steel screw that was implanted over the frontal and parietal lobes. All electrodes were connected to an electrical socket and fixed to the skull with dental cement.
2.3. Semi-rapid kindling procedure
The rats were individually transferred from their home cage to a recording box (30 cm×30cm×30 cm) following a postoperative day 10. A flexible, shielded cable was plugged to the animal’s socket while the animal was moving freely in the recording box. Then, kindling was induced in a semi-rapid manner. Electrical stimulation comprised a 1 ms monophasic square wave of 50 Hz with a 2 s train duration at the afterdischarge threshold. This stimulation paradigm was delivered 6 times daily at inter-train intervals of 20 min. For afterdischarge threshold determination, stimulations were stared at an intensity of 10 µA. The intensities were then increased in increments of 10 µA (with 10 min intervals) until the stimulation could induce the ADs for at least 20 s. This intensity was considered as the afterdischarge threshold and employed for kindling stimulations. The afterdischarge was defined as spikes observed in local field potentials with a frequency of at least 1 Hz and an amplitude of at least twice the baseline activity recorded immediately following stimulation pulses. In this study, the afterdischarge threshold intensity of animals ranged from 20 to 100 μA. Extracellular local field potential signals were boosted and sampled (at 10 kHz) using a PC-based data acquisition system (D3111 Data Acquisition, ScienceBeam Co., Iran) and a homemade software. The progression of kindling was examined by monitoring the behavioral seizure stage and estimating the afterdischarges duration following each stimulation. Behavioral seizures were rated according to the Racine’s standard classification (Racine, 1972): stage 1. facial clonus; stage 2. head nodding; stage 3. forelimb clonus; stage 4. rearing and bilateral forelimb clonus; and stage 5. rearing and falling. When the animals expressed stage 5 seizure over 3 consecutive days, they were regarded as fully kindled.
2.4. LFS application
In LFS-treated groups, LFS was administered at 5 min, 6, 18, and 24 h after the last kindling stimulation. LFS was applied as 4 packages of electrical pulses at 5 min intervals; every package comprised 200 monophasic square wave pulses with 0.1 ms in duration and 1 Hz in frequency. This LFS pattern was similar to our previous study (Ghafouri, Fathollahi, Javan, Shojaei, Asgari, A., & Mirnajafi-Zadeh, 2016). The intensity of pulses for LFS was equal to the afterdischarge threshold for each kindled rat.
2.5. Drug administration
1,3-dimethyl-8-cylclopenthylxanthine (CPT; Sigma, UK), as a selective A1 receptor antagonist, and N6-cyclohexyladenosine (CHA; Sigma, UK), as a selective adenosine A1 receptor agonist, were dissolved in DMSO, and then diluted by adding artificial cerebrospinal fluid (aCSF; composition of aCSF has been mentioned in whole-cell patch-clamp procedure) to the desired concentration. The ultimate concentration of DMSO in solutions was 0.1% and their pH was adjusted to 7.35 - 7.40. The solutions were then sterilized through microfilters (0.2 mm, Minisart, NML, Sartorius, Germany). Drugs were delivered to the left lateral ventricle via a 30-gauge cannula, which was 1 mm below the tip of the guide cannula. CPT at a dose of 2 mM and CHA at a dose of 10 mM were microinjected (1 μl over 1 min) into the left lateral ventricle 3 min before each LFS session in CPT and CHA receiving groups, respectively.
2.6. Whole-cell patch-clamp recording
Rats were killed by decapitation 48 h after the last kindling stimulation under ether anesthesia. Slice preparation and whole-cell patch-clamp recording were performed in the same way as described previously (Shojaei et al., 2014). Briefly, brains were rapidly isolated and the right hemisphere was horizontally sliced with 400 μm in thickness using a vibroslicer (Leica VT 1200s, Leica Microsystems AG, Wetzlar, Germany) in ice-cold cutting artificial cerebrospinal fluid composed of (in mM) 238 sucrose, 2.5 KCl, 1 NaH2PO4, 0.5 CaCl2, 2 MgSO4, 26.2 NaHCO3 and 11 D-glucose. Also, pH ranged from 7.35-7.45 after equilibration with 95% O2 and 5% CO2. Osmolarity was adjusted to 290-300 mOsm. Transverse hippocampal slices (400 μm) were prepared using … .
Slices were immediately transferred to a Gibbs chamber containing ACSF and incubated at 32-35°C for 60 min. ACSF was composed of (in mM) 125 NaCl, 3 KCl, 2 CaCl2, 1.3 MgCl2, 1.25 NaH2PO4, 25 NaHCO3, and 10 D-glucose continuously bubbled with 95% O2 and 5% CO2. The osmolality was 295±5 mOsm and pH was adjusted to 7.3-7.4. Before transferring the slices to the submerged recording chamber, they were kept at room temperature (23-25°C) for a minimum of 20 min. Hippocampal CA1 pyramidal neurons were observed using an upright microscope (Axioskop 2 FS MOT; Carl Zeiss, Germany) equipped with infrared CCD camera (IR-1000, MTI, USA). Recording chamber was continuously superfused with the ACSF at a rate of 3 ml/min.
Whole-cell patch-clamp recording under the current-clamp mode was made from CA1 pyramidal neurons. Recording microelectrodes with a tip resistance of 4-7 MΩ (1.5 mm O.D. and 0.86 mm I.D., Sutter, USA) were pulled from borosilicate glass with a horizontal puller (P-97, Sutter Instrument, USA) in 4 steps and filled with intracellular solution containing (in mM) 115 K-gluconate, 20 KCl, 10 disodium-phosphocreatine, 2 EGTA, 10 HEPES, 2 MgATP and 0.3 Na2GTP. We adjusted pH to 7.25-7.30 and osmolality was set to 290-295 mOsm. Pipette capacitance compensation and bridge balance were carried out and series resistance was compensated by 80%. Signals were recorded by a Multiclamp 700B amplifier and digitized with a Digidata 1440 A/D converter (Molecular Devices, CA, USA). Signals were filtered and digitized at 10 kHz. Electrophysiological analysis was performed using pClamp 10 software.
Measurement of the electrophysiological parameters was started 10 min after the establishment of whole-cell configuration. To estimate the firing properties of neurons, depolarizing current pulses (100 to 200 pA, 50 pA increment, 650 ms) were injected and the number of evoked action potentials (in each depolarizing current step), as well as the onset latency, amplitude of the first spike, adaptation index, instantaneous frequency and post-AHP amplitude (in the 150 pA depolarizing current step) were measured. Also, a depolarizing ramp current (0 - 200 pA during 1000 ms) was injected to calculate the rheobase and utilization time. Action potential amplitude was considered as the voltage difference between the baseline and peak of the first evoked action potential. First spike latency was defined as the time elapsed from the onset of depolarizing current injection to the threshold of the evoked action potential. Instantaneous frequency was defined as the frequency of first two spikes generated in response to applying depolarizing current. The adaptation index of spike frequency was calculated by dividing the average of the last three inter-spike intervals to the average of the first three inter-spike intervals of evoked action potentials.
The post-AHP was measured as the voltage difference between the baseline (before current injection) and the peak of hyperpolarization produced after cessation of depolarizing current injection. The rheobase was considered as the current injected for evoking the first action potential. The delay in firing the first action potential in this situation was measured as utilization time.
2.7. Experimental groups
After the recovery period, animals were divided into seven groups. In the kindled+LFS (KLFS) group (n=3), LFS was administered at 5 min, 6, 18, and 24 h after the last kindling stimulation, and 1 μM ASCF containing 0.1 % DMSO was injected into the lateral ventricle as a vehicle (for 1 min) before each LFS session. In the KLFS+CPT groups (n=3), animals experienced the same procedure; however, they received CPT instead of the vehicle. Animals of the kindled+CHA (n=3) and kindled+CPT (n=3) groups received CHA or CPT, respectively, at the same time after the kindling procedure, LFS was not administrated. In the kindled group (n=3), animals were subjected to only kindling stimulations and in the kindled+Vehicle group (n=3), they received vehicle following kindling acquisition. Animals of the LFS group (n=3) were treated with LFS alone. There were also two control groups, including the sham-operated (n=3) and naïve (non-operated) (n=3) rats. There was no statistically significant difference between the kindled and kindled+vehicle, and also between the sham-operated and naïve groups; therefore, their data were combined into a kindled (n=6) and control (n=6) groups, respectively. Besides, as no statistical difference in various parameters was found between the kindled and kindled+CPT groups, the data of the kindled+CPT group is not shown.
2.8. Statistical analysis
Statistical analysis was carried out using GraphPad Prism software (version 6.01, GraphPad Software, Ca, USA). Normal distribution of data was checked using Shapiro-Wilk and Levene’s test, respectively. The number of evoked action potentials was compared by one-way ANOVA and other parameters were analyzed using a two-way ANOVA (both followed by a Tukey’s post-hoc test). Values are presented as mean±Standard Error of the Mean (SEM) and a p-value of less than 0.05 was considered statistically significant.
3. Results
The electrophysiological properties of CA1 pyramidal neurons were evaluated using whole-cell patch-clamp recording. To characterize the role of adenosine A1 receptors in the effectiveness of LFS on cellular hyperexcitability, the evoked firing activity of pyramidal cells was examined in response to the injection of depolarizing current pulses. A significant difference was observed in the number of action potentials evoked in response to injecting different depolarizing current steps (Figure 1A).
.png)
A two-way ANOVA of the number of evoked action potentials revealed a significant difference in terms of the group (F5,150=35.28, P<0.001) and current intensities (F2,50=115.9, P<0.001). As Figure 1A shows and we reported in our previous studies (Ghotbedin et al., 2013; Moradi Chameh et al., 2015; Shojaei et al., 2014), the excitability of CA1 pyramidal neurons enhanced by kindling stimulations as evidenced by a higher number of action potentials triggered by an injection of 100 to 200 pA depolarizing currents in the kindled group compared with the control group (11.75±1.01 in 100 pA, 18.33±0.90 in 150 pA, and 21.92±0.96 in 200 pA in the kindled group vs. 5.33±0.99 in 100 pA, 11.50±1.08 in 150 pA, and 14.67±1.33 in 200 pA in the control group; P<0.001; n=12 cells in 6 slices from 6 rats; Figure 1A). This parameter indicated a significant reduction due to LFS application in the KLFS group (2.00±1.09 in 100 pA, 6.25±0.77 in 150 pA, and 10.34±0.73 in 200 pA; P<0.001 compared with the kindled group; n=8 cells in 6 slices from 3 rats). Antagonism of adenosine A1 receptors in KLFS+CPT group significantly alleviated the suppressive effect of LFS on the number of evoked action potentials (6.12±0.89 in 100 pA, 12.62±0.10 in 150 pA and 16.87±1.01 in 200 pA; P<0.01 in 100 pA and 150 pA, P<0.05 in 200 pA; n=8 cells in 6 slices from 3 rats). Activation of these receptors following seizure development could mimic the ameliorative effect of LFS and result in lowering the number of fired action potentials in all depolarizing steps (2.37±1.03 in 100 pA; 8.62±1.77 in 150 pA and 14.25±1.90 in 200 pA; P<0.001 compared with the kindled group; n=8 cells in 6 slices from 3 rats). This parameter didn’t show significant changes in the LFS group compared to control (Figure 1A).
A one-way ANOVA showed a significant difference in latency to the first spike among different experimental groups (F5,50=4.20; P<0.01, Figure 1B). In detail, kindling stimulations diminished this latency (from 81.53±20.64 ms in control to 45.22±1.43 ms in kindled group), but we couldn’t find significant difference compare to control (P=0.32; n=12 cells in 6 slices from 6 rats). However, LFS administration to animals of KLFS group increased first spike latency (117.80±13.36 ms; P<0.01 compared to kindled group; n=8 cells in 6 slices from 3 rats). Inhibition of adenosine A1 receptors prevented the effect of LFS on first spike latency (51.34±2.30 ms; P<0.05 compared to KLFS group; n=8 cells in 6 slices from 3 rats). Injection of CHA was also capable to recover this parameter in kindled+CHA group (104.30±20.75 ms; P<0.05 compared to kindled group; n=8 cells in 6 slices from 3 rats). Employment of LFS alone in LFS group had no significant effect on this parameter (55.76±5.04 ms; P=0.78 compared to control) (Figure 1B).
Evaluation of spike accommodation demonstrated a statistically significant difference in adaptation index among experimental groups (F5,46=12.42; P<0.001; Figure 1A). This index showed a considerable reduction in kindled group (1.11±0.02 in kindled vs 1.53±0.05 in control group; P<0.001; n=12 cells in 6 slices from 6 rats). LFS treatment of fully kindled animals in KLFS group could significantly restore this parameter near to control values (1.42±0.09; P<0.01 compared to kindled group; n=6 cells in 6 slices from 3 rats). Blockade of adenosine A1 receptors by CPT in the KLFS+CPT group completely eliminated the effectiveness of LFS on the adaptation index (1.10±0.04; P<0.05 compared to KLFS group; n=8 cells in 6 slices from 3 rats). This parameter significantly enhanced by activation of adenosine A1 receptors in kindled+CHA group (1.46±0.06; P<0.01 compared to kindled group; n=6 cells in 6 slices from 3 rats). It showed a significant reduction by treatment of animals with LFS alone in LFS group (1.20±0.08; P<0.001 compared to control; n=8 cells in 6 slices from 3 rats) (Figure 1A).
We next measured AHP magnitude after a 150 pA current injection (post-AHP). Statistical analysis revealed that AHP had a significantly different amplitude than other experimental groups (F5,50=9.180; P<0.001; Figure 1C). This parameter significantly decreased due to kindling development (from -2.66±0.25 mV in the control group to -1.63±0.19 mV in the kindled group; P<0.05; n=12 cells in 6 slices from 6 rats). However, it completely improved by the LFS application in the KLFS group (-2.78±0.23 mV; P<0.05 compared with the kindled group; n=8 cells in 6 slices from 3 rats). The amplitude of post-AHP in the KLFS+CPT group did not show an alteration following LFS treatment and a significant difference was observed compared with the KLFS group (-1.67±0.15 mV; P<0.05 than the KLFS group; n=8 cells in 6 slices from 3 rats). Examination of this parameter in the kindled+CHA group showed that it recovered by adenosine A1 receptor activation (2.81±0.14 mV; P<0.05 in comparison with the kindled group; n=8 cells in 6 slices from 3 rats). Administration of LFS also significantly decreased post-AHP amplitude in the LFS group (-1.25±0.29 mV; P<0.001 compared with the control; n=8 cells in 6 slices from 3 rats) (Figure 1C).
Estimating the amplitude of the first spike evoked by 150 pA depolarizing current revealed that it declined by kindling stimulations in the kindled group (from 118.90±0.53 mV in control to 109.60±1.17 mV in the kindled group; P<0.001 in comparison with control; n=12 cells in 6 slices from 6 rats, Figure 2A) but LFS restored this parameter in the KLFS group (121.30±0.55 mV; P<0.001 compared with the kindled group; n=8 cells in 6 slices from 3 rats).
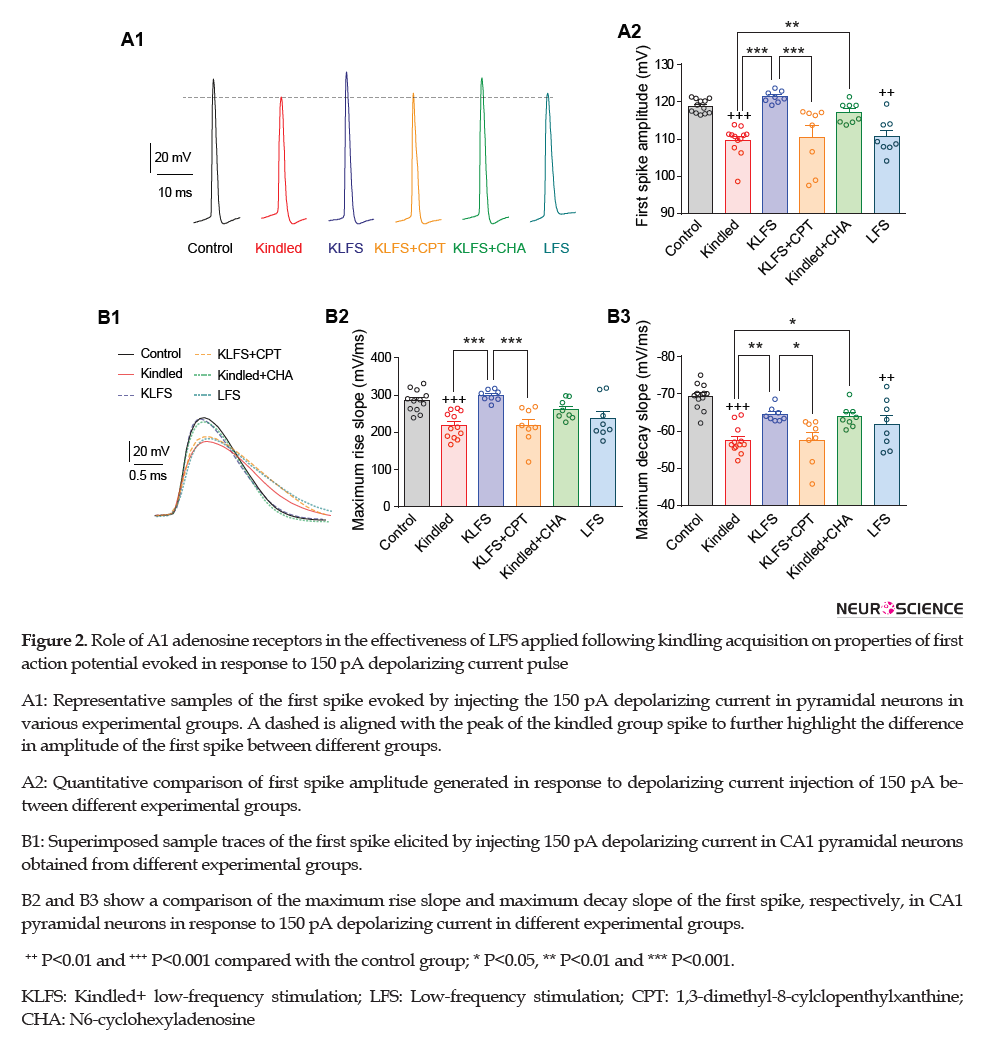
LFS application following CPT injection in the KLFS+CPT group failed to recover kindling-induced decrement of first spike amplitude (110.60±2.97 mV; P<0.001 than the KLFS group; n=8 cells in 6 slices from 3 rats). Activation of adenosine A1 receptors by CHA in the kindled+CHA group could significantly increase this parameter in comparison with the kindled group (117.10±1.00 mV; P<0.01 vs. the kindled group; n=8 cells in 6 slices from 3 rats). Similar to the effect of kindling stimulations, the application of LFS alone also significantly decreased amplitude of first spike (110.70±1.70 mV; P<0.01 compared with the control group; n=8 cells in 6 slices from 3 rats) (Figure 2A).
Maximum rise slope and maximum decay slope of the first action potential were also decreased by kindling stimulations (from 285±8.40 mV/ms in the control group to 219±10 mV/ms in the kindled group for maximum rise slope and form -69±1.00 mV/ms in the control group to -57±1.10 mV/ms in the kindled group for maximum decay slope; P<0.001; n=12 cells in 6 slices from 6 rats; Figure 2B). LFS application in the KLFS group significantly recovered both parameters (299±5.40 mV/ms for maximum rise slope and -64±0.69 mV/ms for maximum decay slope; P<0.001 for maximum rise slope and P<0.01 for maximum decay slope compared with the kindled group; n=8 cells in 6 slices from 3 rats). Administration of CPT and inhibition of adenosine A1 receptors before LFS application in the KLFS+CPT group significantly eliminated the efficacy of LFS in raising these parameters (218±17.00 mV/ms for maximum rise slope (P<0.001) and -58±2.10 mV/ms for maximum decay slope (P<0.05) compared with the KLFS group; n=8 cells in 6 slices from 3 rats). Post-kindling treatment of the animals with CHA and activation of adenosine A1 receptors in the kindled+CHA group significantly increased the maximum decay slope of the first evoked spike than the kindled animals (-64±1.00 mV/ms; P<0.05). Although the enhancement of maximum rise slope in this group was not significant compared with the kindled group, however, no significant difference was observed than the control group (262±17.00 mV/ms; P=0.12 vs. the kindled group and P=0.73 vs. the control group; n=8 cells in 6 slices from 3 rats). LFS application in the LFS group insignificantly declined maximum rise slope and significantly reduced maximum decay slope of the first evoked spike (237±19.00 mV/ms for maximum rise slope (P=0.06) and -62±2.30 mV/ms for maximum decay slope (P<0.01) compared with the KLFS group; n=8 cells in 6 slices from 3 rats) (Figure 2B).
We then measured rheobase and utilization time parameters by applying ramp depolarizing currents. As we reported previously (Ghotbedin et al., 2013; Moradi Chameh, Janahmadi, Semnanian, Shojaei, & Mirnajafi-Zadeh, 2015), both parameters showed a significant reduction following kindling acquisition (form 114.70±9.85 pA in the control group to 79.48±3.99 pA in the kindled group for rheobase (P<0.01), and form 565.40±48.76 ms in the control group to 393.70±77.92 ms in the kindled group for utilization time (P<0.05); n=12 cells in 6 slices from 6 rats; Figure 3).
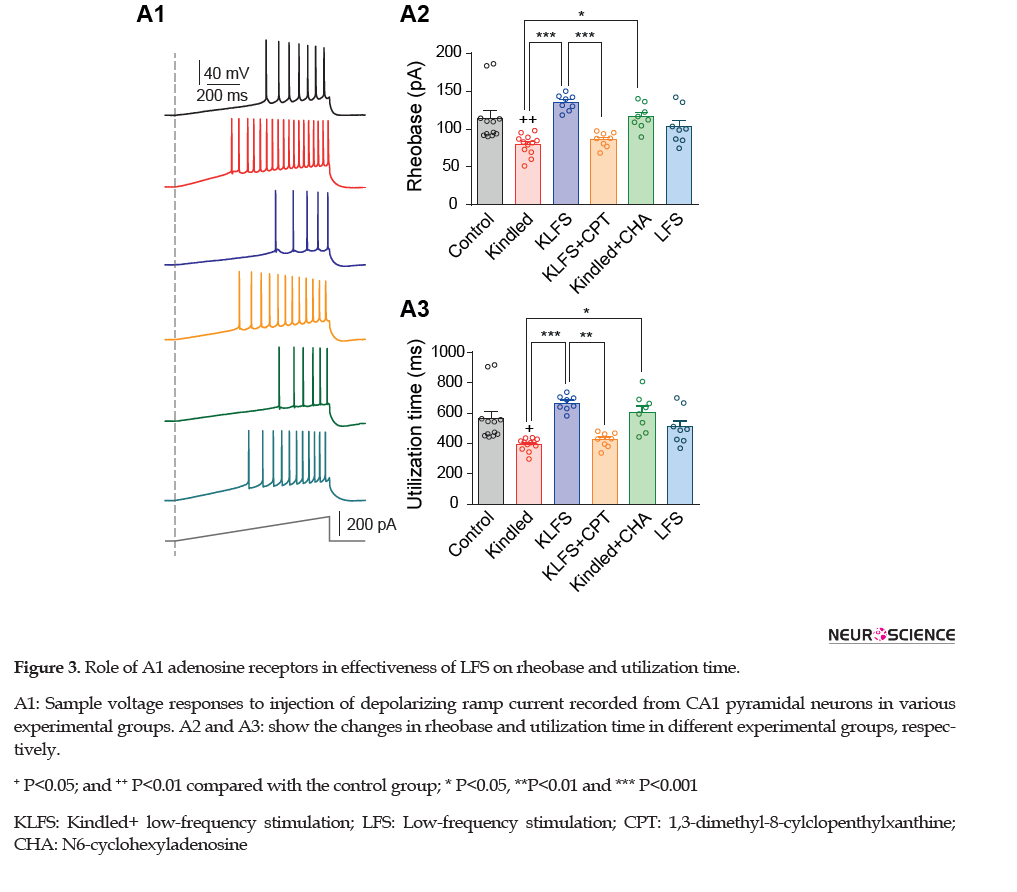
LFS treatment of animals in the KLFS group significantly increased rheobase and utilization time (135.10±3.77 pA for rheobase and 667.60±17.80 ms for utilization time; P<0.001; n=8 cells in 6 slices from 3 rats). CPT injection in the animals of the kindled group significantly eliminated the efficacy of LFS treatment on recovering these parameters (86.13±3.53 pA for rheobase (P<0.001) and 423.80±17.41 ms for utilization time (P<0.01) in the KLFS+CPT group compared with the KLFS group; n=8 cells in 6 slices from 3 rats). CHA administration and activation of adenosine A1 receptors could again mimic LFS effect on these parameters (116±50 pA for rheobase (P<0.01) and 606.40±42.82 ms for utilization time (P<0.001) in kindled+CHA group compared with the kindled group; n=8 cells in 6 slices from 3 rats). The results obtained from the LFS group showed that LFS alone did not affect rheobase (103.50±8.44 pA; P=0.085) and utilization time (509.70±41.70 ms; P=0.86) in comparison with the control group; n=8 cells in 6 slices from 3 rats) (Figure 3).
4. Discussion
In this study, we assessed the role of adenosine A1 receptors in recovering the seizure-induced hyperexcitability of CA1 pyramidal neurons by LFS treatment. The obtained results confirmed that the application of LFS can improve the deleterious effect of kindling on cellular excitability. Also, inhibition of adenosine A1 receptors decreased the effectiveness of LFS in the reduction of cellular excitability in kindled animals. Furthermore, restoring effect of LFS on cellular excitability could be mimicked to a great extent by activation of A1 adenosine receptor. We formerly showed that antiepileptogenic effects of LFS are mediated somehow through the activation of adenosine A1 receptors (Mohammad-Zadeh et al., 2009). In addition, LFS exerted its anticonvulsant effect, in part, by preventing the kindling-induced decrement of A1 receptor gene expression during epileptogenesis (Jahanshahi et al., 2009).
Data obtained from the present study confirmed that LFS could decrease neuronal excitability, indicated by reducing the number of evoked action potentials, extending first spike latency, increasing the adaptation index, post-AHP amplitude, maximum decay slope of the first evoked action potential, rheobase current, and utilization time parameters. Investigating the role of adenosine A1 receptors in these observations revealed that these receptors are involved in restoring the effect of LFS on the number of evoked action potentials.
This parameter is regulated by different voltage- or calcium-dependent outward potassium currents comprising A-type (Lien & Jonas, 2003C; Rudy & McBain, 2001; Storm, 1987; Wang et al., 1998) slowly inactivating D-type (Segal & Barker, 1984; Storm, 1990), slowly activating, non-inactivating M type (Gu, Vervaeke, Hu, & Storm, 2005; Marrion, 1997), large-conductance, calcium-activated (BK) potassium channels (Gu, Vervaeke, & Storm, 2007; Shao, Halvorsrud, Borg-Graham, & Storm, 1999; Storm, 1987) and small-conductance calcium-activated (SK) potassium channels (Engel, Schultens, & Schild,, 1999; Pedarzani et al., 2005). These currents underlie neuronal excitability and the abnormality in their functions leads to increased neuronal excitability and seizure (Brenner et al., 2005; Du et al., 2005; Oliveira et al., 2010; Segal, Barker, 1984; Singh et al., 1998; Wu & Barish, 1999). Therefore, LFS may involve adenosine A1 receptors to affect the activity of one or more types of potassium currents.
For a better clarification, we also assessed the effect of LFS application in the presence of A1 adenosine receptor antagonist (CPT) on maximum decay slope of the first spike, first spike latency, spike adaptation, and post-AHP amplitude in response to a depolarizing current injection. Maximum decay slope and onset latency of the first action potential parameters were hampered to be changed by LFS when adenosine A1 receptors were blocked. Maximum decay slope of the first action potential is dependent on the activity of A-type potassium channels (Bean, 2007; Kim, Wei, & Hoffman, 2005) and first spike latency is mainly determined by the function of both A- and D-type potassium currents (Storm, 1988; Yuan & Chen, 2006). These potassium channels play a pivotal role in the regulation of neuronal excitability (Nakajima, Nakajima, Leonard, & Yamaguchi, 1986; Rogawski, 1985; Segal & Barker, 1984). Based on the findings of this study, it can be suggested that the employed adenosine A1 receptors by LFS can affect A- and D- type potassium currents.
LFS also lost its efficacy on increasing the spike adaptation and post-AHP amplitude when adenosine A1 receptors were blocked. These parameters are mostly dependent on big- and small-conductance (BK and SK) potassium currents (Figenschou, Hu, & Storm, 1996; Gu, Vervaeke, & Storm, 2007). Therefore, the recovery of these currents by LFS may be mediated through adenosine A1 receptors.
The restoring effect of LFS on maximum rise slope of the first evoked action potential, rheobase current, and utilization time was stopped when adenosine A1 receptors were blocked. These parameters are mostly controlled by voltage-gated sodium channels. Considering the important role of these channels in the generation of action potentials (Offord & Catterall, 1989; Waxman, ib-Hajj, Cummins, & Black, 2000), changes in the activity of them by adenosine A1 receptors may also be considered as a possible LFS mechanism of action. In this regard, it has also been shown that the expression of four voltage-gated sodium channel subtypes (NaV1.1, NaV1.2, NaV1.3, and NaV1.6) is elevated in the hippocampus and temporal lobe cortex following epileptic seizures (Berkovic, Mulley, Scheffer, & Petrou, 2006; Blumenfeld et al., 2009; Mulley, Scheffer, Petrou, Dibbens, Berkovic, & Harkin, 2005; Waxman et al., 2000; Xu et al., 2013).
To confirm the possible involvement of adenosine A1 receptors in the ameliorating effect of LFS, we observed that the application of an A1 adenosine receptor agonist (CHA) could mimic the effect of LFS on cellular excitability in kindled animals. Previous studies have demonstrated that the activation of adenosine A1 receptors hyperpolarizes the cells by direct triggering inwardly-rectifying potassium channels (GIRKs) in postsynaptic neurons and decreases cellular excitability (Nicoll, 1988; North, 1989; Rotermund et al., 2018; Thompson, Capogna, & Scanziani, 1993). GIRK1–3 are expressed relatively at high levels by hippocampal CA1 and CA3 pyramidal cells (Drake, Bausch, Milner, & Chavkin, 1997; Karschin, Dissmann, Stühmer, & Karschin, 1996; Liao, Jan, & Jan, 1996; Ponce et al., 1996). Activation of these currents through adenosine A1 receptors inhibits action potential generation in response to depolarizing current injection to CA1 neurons (Hargus, Bertram, & Patel, 2009).
Besides, activation of adenosine A1 receptors can activate calcium-dependent potassium-current flows through the SK channels (Gerber, Greene, Haas, & Stevens, 1989; Haas & Greene, 1984). These channels are controlled by cAMP and underlie the accommodation of firing seen in many hippocampal pyramidal neurons (Marrion & Tavalin, 1998). Furthermore, adenosine reduces the High Voltage-activated (HVA) calcium currents through adenosine A1 receptors in CA1 (Scholz & Miller, 1991) and CA3 (Mogul, Adams, & Fox, 1993) hippocampal pyramidal cells.
It should be noted that the activation of these receptors can also affect synaptic transmission. Adenosine A1 receptors exist at excitatory synapses in the hippocampus (Rebola, Pinheiro, Oliveira, Malva, & Cunha, 2003) and their activation has been demonstrated to hamper excitatory synaptic transmission in CA1 (Brundege & Dunwiddie, 1996) and CA3 pyramidal neurons. Altogether, such mechanisms may be employed by adenosine A1 receptors to affect sodium and potassium currents and reduce the neuronal excitability in kindled animals. However, further studies are required to elucidate this issue in more details.
In conclusion, according to the present data, it may be suggested that LFS involves adenosine A1 receptors to exert its efficacy on neuronal excitability following seizure development in the CA1 area of the hippocampus. The results of this study also confirm the fact that adenosine A1 receptors can be considered as a potential target for drug development against epilepsy.
Ethical Considerations
Compliance with ethical guidelines
All ethical principles were considered in this article.
Funding
This study was financially supported by the Iran National Sciences Foundation (Grant No.: 95000649).
Authors' contributions
All authors contributed equally in preparing all parts of the research.
Conflict of interest
The authors declared no conflict of interest regarding the publication of this article.
References
Ahmadirad, N., Fathollahi, Y., Janahmadi, M., Shojaei, A., Ghasemi, Z., & Barkley, V., et al. (2019). Low-frequency electrical stimulation reduces the impairment in synaptic plasticity following epileptiform activity in rat hippocampal slices through α1, But Not α2, adrenergic receptors. Neuroscience, 406, 176-85. [DOI:10.1016/j.neuroscience.2019.03.007] [PMID]
Alasvand Zarasvand, M., Mirnajafi-Zadeh, J., Fathollahi, Y., & Palizvan, M. R. (2001). Anticonvulsant effect of bilateral injection of N6-cyclohexyladenosine into the CA1 region of the hippocampus in amygdala-kindled rats. Epilepsy Research, 47(1-2), 141-9. [DOI:10.1016/S0920-1211(01)00300-X]
Bean, B. P. (2007). The action potential in mammalian central neurons. Nature Reviews Neuroscience, 8(6), 451-65. [DOI:10.1038/nrn2148] [PMID]
Berkovic, S. F., Mulley, J. C., Scheffer, I. E., & Petrou, S. (2006). Human epilepsies: Interaction of genetic and acquired factors. Trends in Neurosciences, 29(7), 391-7. [DOI:10.1016/j.tins.2006.05.009] [PMID]
Blumenfeld, H., Lampert, A., Klein, J. P., Mission, J., Chen, M. C., & Rivera, M., et al. (2009). Role of hippocampal sodium channel Nav1.6 in kindling epileptogenesis. Epilepsia, 50(1), 44-55. [DOI:10.1111/j.1528-1167.2008.01710.x] [PMID] [PMCID]
Boison, D. (2005). Adenosine and epilepsy: From therapeutic rationale to new therapeutic strategies. Neuroscientist, 11(1), 25-36. [DOI: 10.1177/1073858404269112]
Boison, D. (2013). Role of adenosine in status epilepticus: A potential new target? Epilepsia, 54(s6), 20-2. [DOI:10.1111/epi.12268] [PMID] [PMCID]
Brenner, R., Chen, Q. H., Vilaythong, A., Toney, G. M., Noebels, J. L., & Aldrich, R. W. (2005). BK channel beta4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nature Neuroscience, 8(12), 1752-9. [DOI:10.1038/nn1573] [PMID]
Brundege, J. M., & Dunwiddie, T. V. (1996). Modulation of excitatory synaptic transmission by adenosine released from single hippocampal pyramidal neurons. The Journal of Neuroscience, 16(18), 5603-12. [DOI:10.1523/JNEUROSCI.16-18-05603.1996] [PMID] [PMCID]
Chwalczuk, K., Rubaj, A., Swiader, M., & Czuczwar, S. J. (2008). [Influence of the antagonist of adenosine A1 receptors, 8-cyclopentyl-1 ,3-dipropylxanthine, upon the anticonvulsant activity of antiepileptic drugs in mice (Polish)]. Przegla̧d Lekarski, 65(11), 759-63. [PMID]
de Mendonça, A., & Ribeiro, J. A. (1997). Adenosine and neuronal plasticity. Life Sciences, 60(4-5), 245-51. [DOI:10.1016/S0024-3205(96)00544-9]
Drake, C. T., Bausch, S. B., Milner, T. A., & Chavkin, C. (1997). GIRK1 immunoreactivity is present predominantly in dendrites, dendritic spines, and somata in the CA1 region of the hippocampus. Proceedings of the National Academy of Sciences of the United States of America, 94(3), 1007-12. [DOI:10.1073/pnas.94.3.1007] [PMID] [PMCID]
Du, W., Bautista, J. F., Yang, H., Diez-Sampedro, A., You, S. A., & Wang, L., et al. (2005). Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nature Genetics, 37(7), 733-8. [DOI:10.1038/ng1585] [PMID]
Dunwiddie, T. V., Hoffer, B. J., & Fredholm, B. B. (1981). Alkylxanthines elevate hippocampal excitability. Evidence for a role of endogenous adenosine. Naunyn-Schmiedeberg’s Archives of Pharmacology, 316(4), 326-30. [DOI:10.1007/BF00501365] [PMID]
During, M. J., & Spencer, D. D. (1992). Adenosine: A potential mediator of seizure arrest and postictal refractoriness. Annals of Neurology, 32(5), 618-24. [DOI:10.1002/ana.410320504] [PMID]
Ekonomou, A., Angelatou, F., Vergnes, M., & Kostopoulos, G. (1998). Lower density of A1 adenosine receptors in nucleus reticularis thalami in rats with genetic absence epilepsy. Neuro Report, 9(9), 2135-40. [DOI:10.1097/00001756-199806220-00042] [PMID]
Engel, J. (2001). A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: Report of the ILAE task force on classification and terminology. Epilepsia, 42(6), 796-803. [DOI:10.1046/j.1528-1157.2001.10401.x] [PMID]
Engel, J., Schultens, H. A., & Schild, D. (1999). Small conductance potassium channels cause an activity-dependent spike frequency adaptation and make the transfer function of neurons logarithmic. Biophysical Journal, 76(3), 1310-9. [DOI:10.1016/S0006-3495(99)77293-0]
Esmaeilpour, Kh., Sheibani, V., Shabani, M., & Mirnajafi-Zadeh, J. (2017). Effect of low frequency electrical stimulation on seizure-induced short- and long-term impairments in learning and memory in rats. Physiology & Behavior, 168, 112-21. [DOI:10.1016/j.physbeh.2016.11.001] [PMID]
Fedele, D. E., Li, T., Lan, J. Q., Fredholm, B. B., & Boison, D. (2006). Adenosine A1 receptors are crucial in keeping an epileptic focus localized. Experimental Neurology, 200(1), 184-90. [DOI:10.1016/j.expneurol.2006.02.133] [PMID]
Figenschou, A., Hu, G. Y., & Storm, J. F. (1996). Cholinergic modulation of the action potential in rat hippocampal neurons. The European Journal of Neuroscience, 8(1), 211-9. [DOI:10.1111/j.1460-9568.1996.tb01182.x] [PMID]
Fredholm, B. B., Chen, J. F., Masino, S. A., & Vaugeois, J. M. (2005). Actions of adenosine at its receptors in the CNS: Insights from knockouts and drugs. Annual Review of Pharmacology and Toxicology, 45, 385-412. [DOI:10.1146/annurev.pharmtox.45.120403.095731] [PMID]
Fredholm, B. B., IJzerman, A. P., Jacobson, K. A., Klotz, K. N., & Linden, J. (2001). International :union: of pharmacology. XXV. nomenclature and classification of adenosine receptors. Pharmacological Reviews, 53(4), 527-52. http://pharmrev.aspetjournals.org/content/53/4/527
French, J. A. (2007). Refractory epilepsy: Clinical overview. Epilepsia, 48 (Suppl 1), 3-7. [DOI:10.1111/j.1528-1167.2007.00992.x] [PMID]
Gaito, J., & Gaito, S. T. (1981). The effect of several intertrial intervals on the 1 Hz interference effect. The Canadian Journal of Neurological Sciences, 8(1), 61-5. [DOI:10.1017/S0317167100042864] [PMID]
Gasior, M., Borowicz, K., Kleinrok, Z. S., & Czuczwar, S. J. (1996). Chronic caffeine and the anticonvulsant potency of antiepileptic drugs against maximal electroshock. Pharmacology Biochemistry and Behavior, 54(4), 639-44. [DOI:10.1016/0091-3057(95)02222-8]
Gerber, U., Greene, R. W., Haas, H. L., & Stevens, D. R. (1989). Characterization of inhibition mediated by adenosine in the hippocampus of the rat in vitro. The Journal of Physiology, 417, 567-78. [DOI:10.1113/jphysiol.1989.sp017819] [PMID] [PMCID]
Ghafouri, S., Fathollahi, Y., Javan, M., Shojaei, A., Asgari, A., & Mirnajafi-Zadeh, J. (2016). Effect of low frequency stimulation on impaired spontaneous alternation behavior of kindled rats in Y-maze test. Epilepsy Research, 126, 37-44. [DOI:10.1016/j.eplepsyres.2016.06.010] [PMID]
Gharib, A. R., Sayyahi, Z., Komaki, A. R., Barkley, V., Sarihi, A., & Mirnajafi-Zadeh, J. (2018). The role of 5-HT1A receptors of hippocampal CA1 region in anticonvulsant effects of low-frequency stimulation in amygdala kindled rats. Physiology & Behavior, 196, 119-25. [DOI:10.1016/j.physbeh.2018.08.025] [PMID]
Ghasemi, Z., Naderi, N., Shojaei, A., Raoufy, M. R., Ahmadirad, N., & Barkley, V., et al. (2019). The inhibitory effect of different patterns of low frequency stimulation on neuronal firing following epileptiform activity in rat hippocampal slices. Brain Research, 1706, 184-95. [DOI:10.1016/j.brainres.2018.11.012] [PMID]
Ghorbani, P., Mohammad-Zadeh, M., Mirnajafi-Zadeh, J., & Fathollahi, Y. (2007). Effect of different patterns of low-frequency stimulation on piriform cortex kindled seizures. Neuroscience Letters, 425(3), 162-6. [DOI:10.1016/j.neulet.2007.08.023] [PMID]
Ghotbedin, Z., Janahmadi, M., Mirnajafi-Zadeh, J., Behzadi, G., & Semnanian, S. (2013). Electrical low frequency stimulation of the kindling site preserves the electrophysiological properties of the rat hippocampal CA1 pyramidal neurons from the destructive effects of amygdala kindling: The basis for a possible promising epilepsy therapy. Brain Stimulation, 6(4), 515-23. [DOI:10.1016/j.brs.2012.11.001] [PMID]
Goodman, J. H., Berger, R. E., & Tcheng, T. K. (2005). Preemptive low-frequency stimulation decreases the incidence of amygdala-kindled seizures. Epilepsia, 46(1), 1-7. [DOI:10.1111/j.0013-9580.2005.03804.x] [PMID]
Gu, N., Vervaeke, K., & Storm, J. F. (2007). BK potassium channels facilitate high-frequency firing and cause early spike frequency adaptation in rat CA1 hippocampal pyramidal cells. The Journal of Physiology, 580(Pt 3), 859-82. [DOI:10.1113/jphysiol.2006.126367] [PMID] [PMCID]
Gu, N., Vervaeke, K., Hu, H., & Storm, J. F. (2005). Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. The Journal of Physiology, 566(Pt 3), 689-715. [DOI:10.1113/jphysiol.2005.086835] [PMID] [PMCID]
Haas, H. L., & Greene, R. W. (1984). Adenosine enhances afterhyperpolarization and accommodation in hippocampal pyramidal cells. Pflügers Archiv, 402(3), 244-7. [DOI:10.1007/BF00585506] [PMID]
Hargus, N. J., Bertram, E. H., & Patel, M. K. (2009). Adenosine A1 receptors presynaptically modulate excitatory synaptic input onto subiculum neurons. Brain Research, 1280, 60-8. [DOI:10.1016/j.brainres.2009.05.027] [PMID] [PMCID]
Hinterkeuser, S., Schröder, W., Hager, G., Seifert, G., Blümcke, I., & Elger, C. E., et al. (2000). Astrocytes in the hippocampus of patients with temporal lobe epilepsy display changes in potassium conductances. The European Journal of Neuroscience, 12(6), 2087-96. [DOI:10.1046/j.1460-9568.2000.00104.x] [PMID]
Hosseinmardi, N., Mirnajafi-Zadeh, J., Fathollahi, Y., & Shahabi, P. (2007). The role of adenosine A1 and A2A receptors of entorhinal cortex on piriform cortex kindled seizures in rats. Pharmacological Research, 56(2), 110-7. [DOI:10.1016/j.phrs.2007.04.011] [PMID]
Jahanshahi, A., Mirnajafi-Zadeh, J., Javan, M., Mohammad-Zadeh, M., & Rohani, R. (2009). The antiepileptogenic effect of electrical stimulation at different low frequencies is accompanied with change in adenosine receptors gene expression in rats. Epilepsia, 50(7), 1768-79. [DOI:10.1111/j.1528-1167.2009.02088.x] [PMID]
Karschin, C., Dissmann, E., Stühmer, W., & Karschin, A. (1996). IRK(1-3) and GIRK(1-4) inwardly rectifying K+ channel mRNAs are differentially expressed in the adult rat brain. The Journal of Neuroscience, 16(11), 3559-70. [DOI:10.1523/JNEUROSCI.16-11-03559.1996] [PMID] [PMCID]
Kim, J., Wei, D. S., & Hoffman, D. A. (2005). Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones. The Journal of Physiology, 569(Pt 1), 41-57. [DOI:10.1113/jphysiol.2005.095042] [PMID] [PMCID]
Kinoshita, M., Ikeda, A., Matsumoto, R., Begum, T., Usui, K., & Yamamoto, J., et al. (2004). Electric stimulation on human cortex suppresses fast cortical activity and epileptic spikes. Epilepsia, 45(7), 787-91. [DOI:10.1111/j.0013-9580.2004.60203.x] [PMID]
Kwan, P., & Brodie, M. J. (2000). Early identification of refractory epilepsy. The New England Journal of Medicine, 342(5), 314-9. [DOI:10.1056/NEJM200002033420503] [PMID]
Liao, Y. J., Jan, Y. N., & Jan, L. Y. (1996). Heteromultimerization of G-protein-gated inwardly rectifying K+ channel proteins GIRK1 and GIRK2 and their altered expression in weaver brain. The Journal of Neuroscience, 16(22), 7137-50. [DOI:10.1523/JNEUROSCI.16-22-07137.1996] [PMID] [PMCID]
Lien, C. C., & Jonas, P. (2003). Kv3 potassium conductance is necessary and kinetically optimized for high-frequency action potential generation in hippocampal interneurons. The Journal of Neuroscience, 23(6), 2058-68. [DOI:10.1523/JNEUROSCI.23-06-02058.2003] [PMID] [PMCID]
Lin, J. S., Lew, S. M., Marcuccilli, C. J., Mueller, W. M., Matthews, A. E., & Koop, J. I., et al. (2011). Corpus callosotomy in multistage epilepsy surgery in the pediatric population. Journal of Neurosurgery. Pediatrics, 7(2), 189-200. [DOI:10.3171/2010.11.PEDS10334] [PMID]
Malhotra, J., & Gupta, Y. K. (1997). Effect of adenosine receptor modulation on pentylenetetrazole-induced seizures in rats. British Journal of Pharmacology, 120(2), 282-8. [DOI:10.1038/sj.bjp.0700869] [PMID] [PMCID]
Marrion, N. V. (1997). Control of M-current. Annual Review of Physiology, 59, 483-504. [DOI:10.1146/annurev.physiol.59.1.483] [PMID]
Marrion, N. V., & Tavalin, S. J. (1998). Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature, 395(6705), 900-5. [DOI:10.1038/27674] [PMID]
Mogul, D. J., Adams, M. E., & Fox, A. P. (1993). Differential activation of adenosine receptors decreases N-type but potentiates P-type Ca2+ current in hippocampal CA3 neurons. Neuron, 10(2), 327-34. [DOI:10.1016/0896-6273(93)90322-I]
Mohammad-Zadeh, M., Mirnajafi-Zadeh, J., Fathollahi, Y., Javan, M., Ghorbani, P., & Sadegh, M., et al. (2007). Effect of low frequency stimulation of perforant path on kindling rate and synaptic transmission in the dentate gyrus during kindling acquisition in rats. Epilepsy Research, 75(2-3), 154-61. [DOI:10.1016/j.eplepsyres.2007.05.003] [PMID]
Mohammad-Zadeh, M., Mirnajafi-Zadeh, J., Fathollahi, Y., Javan, M., Jahanshahi, A., & Noorbakhsh, S. M., et al. (2009). The role of adenosine A1 receptors in mediating the inhibitory effects of low frequency stimulation of perforant path on kindling acquisition in rats. Neuroscience, 158(4), 1632-43. [DOI:10.1016/j.neuroscience.2008.11.008] [PMID]
Moradi Chameh, H., Janahmadi, M., Semnanian, S., Shojaei, A., & Mirnajafi-Zadeh, J. (2015). Effect of low frequency repetitive transcranial magnetic stimulation on kindling-induced changes in electrophysiological properties of rat CA1 pyramidal neurons. Brain Research, 1606, 34-43. [DOI:10.1016/j.brainres.2015.02.023] [PMID]
Mulley, J. C., Scheffer, I. E., Petrou, S., Dibbens, L. M., Berkovic, S. F., & Harkin, L. A. (2005). SCN1A mutations and epilepsy. Human Mutation, 25(6), 535-42. [DOI:10.1002/humu.20178] [PMID]
Nakajima, Y., Nakajima, S., Leonard, R. J., & Yamaguchi, K. (1986). Acetylcholine raises excitability by inhibiting the fast transient potassium current in cultured hippocampal neurons. Proceedings of the National Academy of Sciences of the United States of America, 83(9), 3022-6. [DOI:10.1073/pnas.83.9.3022] [PMID] [PMCID]
Nicoll, R. A. (1988). The coupling of neurotransmitter receptors to ion channels in the brain. Science, 241(4865), 545-51. [DOI:10.1126/science.2456612] [PMID]
North, R. A. (1989). Twelfth Gaddum memorial lecture. Drug receptors and the inhibition of nerve cells. British Journal of Pharmacology, 98(1), 13-28. [DOI:10.1111/j.1476-5381.1989.tb16855.x] [PMID] [PMCID]
Offord, J., & Catterall, W. A. (1989). Electrical activity, cAMP, and cytosolic calcium regulate mRNA encoding sodium channel alpha subunits in rat muscle cells. Neuron, 2(5), 1447-52. [DOI:10.1016/0896-6273(89)90190-6]
Oliveira, M. S., Skinner, F., Arshadmansab, M. F., Garcia, I., Mello, C. F., & Knaus, H. G., et al. (2010). Altered expression and function of small-conductance (SK) Ca2+-activated K+ channels in pilocarpine-treated epileptic rats. Brain Research, 1348, 187-99. [DOI:10.1016/j.brainres.2010.05.095] [PMID] [PMCID]
Paxinos, G., & Watson, C. (2009). The rat brain in stereotaxic coordinates. 6th Ed. Amsterdam: Elsevier. https://books.google.com/books?id=sQNfmwEACAAJ&dq
Pedarzani, P., McCutcheon, J. E., Rogge, G., Jensen, B. S., Christophersen, P., & Hougaard, C., et al. (2005). Specific enhancement of SK channel activity selectively potentiates the afterhyperpolarizing current IAHP and modulates the firing properties of hippocampal pyramidal neurons. The Journal of Biological Chemistry, 280(50), 41404-11. [DOI:10.1074/jbc.M509610200] [PMID]
Ponce, A., Bueno, E., Kentros, C., de Miera, E. V. S., Chow, A., & Hillman, D., et al. (1996). G-protein-gated inward rectifier K+ channel proteins (GIRK1) are present in the soma and dendrites as well as in nerve terminals of specific neurons in the brain. The Journal of Neuroscience, 16(6), 1990-2001. [DOI:10.1523/JNEUROSCI.16-06-01990.1996] [PMID] [PMCID]
Racine, R. J. (1972). [Modification of seizure activity by electrical stimulation: II. Motor seizure (French)]. Electroencephalography and Clinical Neurophysiology, 32(3), 281-94. [DOI:10.1016/0013-4694(72)90177-0]
Rebola, N., Pinheiro, P. C., Oliveira, C. R., Malva, J. O., & Cunha, R. A. (2003). Subcellular localization of adenosine A(1) receptors in nerve terminals and synapses of the rat hippocampus. Brain Research, 987(1), 49-58. [DOI:10.1016/S0006-8993(03)03247-5]
Ribeiro, J. A. (1995). Purinergic inhibition of neurotransmitter release in the central nervous system. Pharmacology & Toxicology, 77(5), 299-305. [DOI:10.1111/j.1600-0773.1995.tb01031.x] [PMID]
Ribeiro, J. A., Sebastiao, A. M., & de Mendonca, A. (2003). Participation of adenosine receptors in neuroprotection. Drug News & Perspectives, 16(2), 80-6. [DOI:10.1358/dnp.2003.16.2.740246] [PMID]
Rogawski, M. A. (1985). The A-current: How ubiquitous a feature of excitable cells is it? Trends in Neurosciences, 8, 214-9. [DOI:10.1016/0166-2236(85)90082-7]
Rotermund, N., Winandy, S., Fischer, T., Schulz, K., Fregin, T., & Alstedt, N., et al. (2018). Adenosine A1 receptor activates background potassium channels and modulates information processing in olfactory bulb mitral cells. The Journal of Physiology, 596(4), 717-33. [DOI:10.1113/JP275503] [PMID] [PMCID]
Rudy, B., & McBain, C. J. (2001). Kv3 channels: Voltage-gated K+ channels designed for high-frequency repetitive firing. Trends in Neurosciences, 24(9), 517-26. [DOI:10.1016/S0166-2236(00)01892-0]
Scholz, K. P., & Miller, R. J. (1991). Analysis of adenosine actions on Ca2+ currents and synaptic transmission in cultured rat hippocampal pyramidal neurones. The Journal of Physiology, 435, 373-93. [DOI:10.1113/jphysiol.1991.sp018515] [PMID] [PMCID]
Segal, M., & Barker, J. L. (1984). Rat hippocampal neurons in culture: Potassium conductances. Journal of Neurophysiology, 51(6), 1409-33. [DOI:10.1152/jn.1984.51.6.1409] [PMID]
Segal, M., Rogawski, M. A., & Barker, J. L. (1984). A transient potassium conductance regulates the excitability of cultured hippocampal and spinal neurons. The Journal of Neuroscience, 4(2), 604-9. [DOI:10.1523/JNEUROSCI.04-02-00604.1984] [PMID] [PMCID]
Shahpari, M., Mirnajafi-Zadeh, J., Firoozabadi, S. M. P., & Yadollahpour, A. (2012). Effect of low-frequency electrical stimulation parameters on its anticonvulsant action during rapid perforant path kindling in rat. Epilepsy Research, 99(1-2), 69-77. [DOI:10.1016/j.eplepsyres.2011.10.023] [PMID]
Shao, L. R., Halvorsrud, R., Borg-Graham, L., & Storm, J. F. (1999). The role of BK-type Ca2+-dependent K+ channels in spike broadening during repetitive firing in rat hippocampal pyramidal cells. The Journal of Physiology, 521(Pt 1), 135-46. [DOI:10.1111/j.1469-7793.1999.00135.x] [PMID] [PMCID]
Shojaei, A., Semnanian, S., Janahmadi, M., Moradi-Chameh, H., Firoozabadi, S. M., & Mirnajafi-Zadeh, J. (2014). Repeated transcranial magnetic stimulation prevents kindling-induced changes in electrophysiological properties of rat hippocampal CA1 pyramidal neurons. Neuroscience, 280, 181-92. [DOI:10.1016/j.neuroscience.2014.09.022] [PMID]
Siebel, A. M., Menezes, F. P., Capiotti, K. M., Kist, L. W., da Costa Schaefer, I., & Frantz, J. Z., et al. (2015). Role of adenosine signaling on pentylenetetrazole-induced seizures in zebrafish. Zebrafish, 12(2), 127-36. [DOI:10.1089/zeb.2014.1004] [PMID] [PMCID]
Singh, N. A., Charlier, C., Stauffer, D., DuPont, B. R., Leach, R. J., & Melis, R., et al. (1998). A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nature Genetics, 18(1), 25-9. [DOI:10.1038/ng0198-25] [PMID]
Storm, J. F. (1987). Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. The Journal of Physiology, 385, 733-59. [DOI:10.1113/jphysiol.1987.sp016517] [PMID] [PMCID]
Storm, J. F. (1988). Temporal integration by a slowly inactivating K+ current in hippocampal neurons. Nature, 336(6197), 379-81. [DOI:10.1038/336379a0] [PMID]
Storm, J. F. (1990). Potassium currents in hippocampal pyramidal cells. In: J. Storm-Mathisen, J. Zimmer, O. P. Ottersen (Eds.). Understanding the brain through the hippocampus the hippocampal region as a model for studying brain structure and function, progress in brain research (pp. 161-187). Vol. 83. Amsterdam: Elsevier Science Publishers. [DOI:10.1016/S0079-6123(08)61248-0]
Temkin, N. R. (2009). Preventing and treating posttraumatic seizures: The human experience. Epilepsia, 50 (Suppl 2), 10-3. [DOI:10.1111/j.1528-1167.2008.02005.x] [PMID]
Thompson, S. M., Capogna, M., & Scanziani, M. (1993). Presynaptic inhibition in the hippocampus. Trends in Neurosciences, 16(6), 222-7. [DOI:10.1016/0166-2236(93)90160-N]
Wang, L. Y., Gan, L., Forsythe, I. D., & Kaczmarek, L. K. (1998). Contribution of the Kv3.1 potassium channel to high-frequency firing in mouse auditory neurones. The Journal of Physiology, 509(Pt 1), 183-94. [DOI:10.1111/j.1469-7793.1998.183bo.x] [PMID] [PMCID]
Waxman, S. G., Dib-Hajj, S., Cummins, T. R., & Black, J. A. (2000). Sodium channels and their genes: Dynamic expression in the normal nervous system, dysregulation in disease states. Brain Research, 886(1-2), 5-14. [DOI:10.1016/S0006-8993(00)02774-8]
Wu, R. L., & Barish, M. E. (1999). Modulation of a slowly inactivating potassium Current,/D, by metabotropic glutamate receptor activation in cultured hippocampal pyramidal neurons. The Journal of Neuroscience, 19(16), 6825-37. [DOI:10.1523/JNEUROSCI.19-16-06825.1999] [PMID] [PMCID]
Xu, X., Guo, F., Lv, X., Feng, R., Min, D., & Ma, L., et al. (2013). Abnormal changes in voltage-gated sodium channels NaV1.1, NaV1.2, NaV1.3, NaV1.6 and in calmodulin/calmodulin-dependent protein kinase II, within the brains of spontaneously epileptic rats and tremor rats. Brain Research Bulletin, 96, 1-9. [DOI:10.1016/j.brainresbull.2013.04.003] [PMID]
Yamamoto, J., Ikeda, A., Kinoshita, M., Matsumoto, R., Satow, T., & Takeshita, K., et al. (2006). Low-frequency electric cortical stimulation decreases interictal and ictal activity in human epilepsy. Seizure, 15(7), 520-7. [DOI:10.1016/j.seizure.2006.06.004] [PMID]
Yamamoto, J., Ikeda, A., Satow, T., Takeshita, K., Takayama, M., & Matsuhashi, M., et al. (2002). Low-frequency electric cortical stimulation has an inhibitory effect on epileptic focus in mesial temporal lobe epilepsy. Epilepsia, 43(5), 491-5. [DOI:10.1046/j.1528-1157.2002.29001.x] [PMID]
Young, D., & Dragunow, M. (1994). Status epilepticus may be caused by loss of adenosine anticonvulsant mechanisms. Neuroscience, 58(2), 245-61. [DOI:10.1016/0306-4522(94)90032-9]
Yuan, L. L., & Chen, X. (2006). Diversity of potassium channels in neuronal dendrites. Progress in Neurobiology, 78(6), 374-89. [DOI:10.1016/j.pneurobio.2006.03.003] [PMID]
Zeraati, M., Mirnajafi-Zadeh, J., Fathollahi, Y., Namvar, S., & Rezvani, M. E. (2006). Adenosine A1 and A2A receptors of hippocampal CA1 region have opposite effects on piriform cortex kindled seizures in rats. Seizure, 15(1), 41-8. [DOI:10.1016/j.seizure.2005.10.006] [PMID]
Zhou, Q., Zhu, S., Guo, Y., Lian, L., Hu, Q., & Liu, X., et al. (2018). Adenosine A1 receptors play an important protective role against cognitive impairment and long-term potentiation inhibition in a pentylenetetrazol mouse model of epilepsy. Molecular Neurobiology, 55(4), 3316-27. [DOI:10.1007/s12035-017-0571-x] [PMID]
Received: 2019/02/17 | Accepted: 2019/07/20 | Published: 2020/05/1
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
