

Volume 10, Issue 4 (July & August 2019)
BCN 2019, 10(4): 281-304 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Sadighparvar S, Tale F, Shahabi P, Naderi S, Ghaderi Pakdel F. The Response of Ventral Tegmental Area Dopaminergic Neurons to Bupropion: Excitation or Inhibition?. BCN 2019; 10 (4) :281-304
URL: http://bcn.iums.ac.ir/article-1-1044-en.html
URL: http://bcn.iums.ac.ir/article-1-1044-en.html
Shirin Sadighparvar1 
 , Fereshteh Tale2 , Parviz Shahabi3 , Somayyeh Naderi4 , Firouz Ghaderi Pakdel *5
, Fereshteh Tale2 , Parviz Shahabi3 , Somayyeh Naderi4 , Firouz Ghaderi Pakdel *5
, Fereshteh Tale2 , Parviz Shahabi3 , Somayyeh Naderi4 , Firouz Ghaderi Pakdel *5
1- Neurophysiology Research Center, Urmia University of Medical Sciences, Urmia, Iran.; Department of Physiology, School of Medicine, Urmia University of Medical Sciences, Urmia, Iran.
2- Department of Physiology, Faculty of Medicine, Urmia University of Medical Sciences, Urmia, Iran
3- Neuroscience Research Center, Tabriz University of Medical Sciences, Tabriz, Iran.
4- Reproductive Health Research Center, Urmia University of Medical Sciences, Urmia, Iran.
5- Neurophysiology Research Center, Urmia University of Medical Sciences, Urmia, Iran.; Reproductive Health Research Center, Urmia University of Medical Sciences, Urmia, Iran.
2- Department of Physiology, Faculty of Medicine, Urmia University of Medical Sciences, Urmia, Iran
3- Neuroscience Research Center, Tabriz University of Medical Sciences, Tabriz, Iran.
4- Reproductive Health Research Center, Urmia University of Medical Sciences, Urmia, Iran.
5- Neurophysiology Research Center, Urmia University of Medical Sciences, Urmia, Iran.; Reproductive Health Research Center, Urmia University of Medical Sciences, Urmia, Iran.
Full-Text [PDF 2689 kb]
| Abstract (HTML)
Full-Text:
1. Introduction
The monoamine hypothesis of depression is a crucial pharmacologic issue for depression based on tricyclic antidepressants and monoamine oxidase inhibitors effects. These drugs mimic serotonin, norepinephrine, and or dopamine. These property has directed research studies to evaluate the role of biogenic monoamine neurotransmitters in depression (Schildkraut, 1967). Dopamine insufficiencies and or deficiencies associated with depression along with endocrine disorders (Diehl & Gershon, 1992). There is some clinical evidence on the role of midbrain dopamine, Ventral Tegmental Area (VTA) in depression (Brown & Gershon, 1993), but it could also be involved in motivation, addiction, and psychosis. Increase in the dopamine in the VTA by Monoamine Oxidase Inhibitors (MAOIs) and or dopamine reuptake inhibitors provide well-known pharmacologic agents to alleviate depression (Randrup & Braestrup, 1977; Sampson, Willner, & Muscat, 1991; Willner, 1983).
The monoamine hypothesis of depression is a crucial pharmacologic issue for depression based on tricyclic antidepressants and monoamine oxidase inhibitors effects. These drugs mimic serotonin, norepinephrine, and or dopamine. These property has directed research studies to evaluate the role of biogenic monoamine neurotransmitters in depression (Schildkraut, 1967). Dopamine insufficiencies and or deficiencies associated with depression along with endocrine disorders (Diehl & Gershon, 1992). There is some clinical evidence on the role of midbrain dopamine, Ventral Tegmental Area (VTA) in depression (Brown & Gershon, 1993), but it could also be involved in motivation, addiction, and psychosis. Increase in the dopamine in the VTA by Monoamine Oxidase Inhibitors (MAOIs) and or dopamine reuptake inhibitors provide well-known pharmacologic agents to alleviate depression (Randrup & Braestrup, 1977; Sampson, Willner, & Muscat, 1991; Willner, 1983).
The VTA comprises of dopamine, Gamma-Aminobutyric Acid (GABAergic), and glutamate-releasing neurons, most of which are dopaminergic and GABAergic (Holly & Miczek, 2016; Nair-Roberts et al., 2008; Yamaguchi, Wang, Li, Ng, & Morales, 2011). Two primary afferents of the VTA; mesolimbic and mesocortical systems, originate from nucleus accumbens, amygdala, hippocampus, prefrontal cortex, and limbic system (Albanese & Minciacchi, 1983; Fallon, 1988; Le Moal & Simon, 1991). The efferents of the VTA send back to the nucleus accumbens to produce modulatory loop (Spanagel, Herz, & Shippenberg, 1992). Enhancing the synaptic dopamine availability increases the postsynaptic responses is associated with depression pharmacotherapy, and for this reason, the agents with elevated dopamine and or other monoamines have profound antidepression outcomes.
Reuptake inhibition of the multi-neurotransmitters has provided a novel and effective generation of antidepressants, such as bupropion (Dremencov et al., 2004; Dremencov et al., 2005; Nutt et al., 2006). Bupropion (Wellbutrin) that is recently re-marketed in the USA and other regions exerts its antidepressant effect by blocking the reuptake of dopamine. Bupropion has a weak inhibitory effect on norepinephrine reuptake, too (Horst & Preskorn, 1998). The confirmed anti-smoke effects of bupropion are believed to antagonize the nicotinic acetylcholine receptors (nAchRs) (Taeron, 2002). Although it is reported that bupropion, primarily blocks the reuptake of both dopamine and norepinephrine, the mechanism of its action has not been fully elucidated (Ascher et al., 1995). The dual effect of bupropion on the dopamine and norepinephrine reuptakes has changed the treatment of specific symptoms of depression, cognitive, and fatigue associated with selective monoamine inhibitors and selective serotonin reuptake inhibitors (SSRIs) (Fabre, Brodie, Garver, & Zung,1983; Green, 1997). Bupropion also has less risk of sexual dysfunction compared to SSRIs (Pereira, Arias-Carrion, Machado, Nardi, & Silva, 2014).
Although bupropion primarily inhibits the dopamine reuptake, its precise mechanism of action is still unknown because of differences in its in vivo and in vitro effects. The in vitro potency of norepinephrine reuptake inhibition of bupropion is about twice than that in vivo (Ascher et al., 1995).
The duration of its administration has different effects on norepinephrine-releasing neurons. In acute systemic application, it can reduce norepinephrine releasing firing rates, but its sustained administration can recover the baseline firing rate (Ghanbari, El Mansari, & Blier, 2010). This effect is suggested due to the somatodendritic α2-adrenoceptors activation (Dong & Blier, 2001). Besides its action on monoamines, a low amount of bupropion can attenuate nicotine-evoked dopamine release in striatum in slice preparations (Mansvelder, Fagen, Chang, Mitchum, & McGehee, 2007; Sidhpura, Redfern, Rowley, Heal, & Wonnacott, 2007). Nicotine can induce significantly large somatic responses in the neurons (Klink, de Kerchove d’Exaerde, Zoli, & Changeux, 2001; Wooltorton, Pidoplichko, Broide, & Dani, 2003) via alpha7-nicotinic receptors and modulate nicotine-induced reinforcement and extracellular dopamine in the mesolimbic system (Besson et al., 2012). The mechanism of the bupropion action on the VTA dopaminergic neurons is not well-known due to the presence of different dopamine receptors and their various locations and destinations pre- and postsynaptically.
Our recent studies showed that acute systemic infusion of bupropion could decrease formalin-induced pain behavior in rats (Naderi, Pakdel, Osalou, & Cankurt, 2014). Microinfusion of the bupropion into the locus coeruleus nuclease could decrease formalin-induced pain response (Jahanbani et al., 2014). The intracerebroventricular application of the bupropion revealed that it could reduce the spontaneous firing of locus coeruleus neurons dose-dependently (Pakdel et al., 2014). The effect of the bupropion on the dendritic adrenergic receptors has been postulated as a critical factor for these actions. It is reported that neither 2 nor 14 days of bupropion (30 mg/kg/d) administration alter the firing and burst activity of dopamine neurons (El Mansari, Ghanbari, Janssen, & Blier, 2008), but there are other reports about increase in firing rate with 14 days administration of bupropion at two 10 and 20 mg/kg/d doses (West & Weiss, 2011). Cooper et al. revealed that the acute systemic infusion of bupropion could inhibit the firing of the VTA dopaminergic neurons (Cooper et al., 1994).
Bupropion can also act on the VTA dopaminergic neurons by VTA GABAergic neurons. The microinfusion of the bupropion could inhibit the neuronal firing of the putative VTA GABAergic neurons dose-dependently (Amirabadi et al., 2014). We hypothesized that the critical neuronal control of the VTA dopaminergic neurons is the GABA neurotransmission and because there is no evidence about the direct effect of bupropion on VTA dopaminergic neurons, this study was designed to test the acute effects of bupropion via intracerebroventricular and microiontophoretic applications on the dopaminergic neuronal firing rates of the VTA.
2. Methods
2.1. Ethical approval
The committee for biomedical research ethics approval reviewed the procedure, experiments, protocols, and all guidelines for the care and use of experimental animals. The Animal Laboratory Center of Urmia University of Medical Sciences (ALCUUMS) supervised precisely over all ethical issues. All experimental procedures and protocols were approved by the Urmia Medical Science Research Ethics Committee (UMSREC). The ethical issues were performed in accordance with the National Institute of Health (NIH) guide for the care and use of laboratory animals.
2.2. Study animals
Healthy male Wistar rats of 250-280 g weight (Pasteur Institute, Tehran, Iran) were housed (three on a cage) at a 12/12 h light/dark cycle (7:00 AM to 7:00 PM). The ambient temperature was controlled (22±2°C), and the food and water were at labium. The two intracerebroventricular and microiontophoretic categories of animals were divided into 14 groups (5 per each subgroup) according to the bupropion application with the sham and control groups too. Apart from the control and sham groups, in the first category, the animals received 1, 0.5, 0.1, 0.01, 0.001, and 0.0001 mol of intracerebroventricular bupropion (5 μL/3 min). In the second category, bupropion was ejected by -50, -150, -300, and -500 nA electrical currents (1 mol of bupropion solution, pH=4.5, 5min) with the control and sham subgroups. Briefly, animals were anesthetized by urethane (1.2 g/kg) initially, and the booster dose (~ 15% to 25% of the initial dose) was used when there were any discomfort signs. The core body temperature was continuously monitored and maintained at ~37°C during the entire experiment. The stereotaxic coordinations of bregma and interaural landmarks were determined for each rat, and the location of recording and injecting electrodes were exposed according to the Paxinos and Watson stereotaxic rat brain atlas (Paxinos & Watson, 2007).
2.3. VTA multiple electrophysiological recording
Multiunit extracellular electrophysiological activity of the VTA dopaminergic neurons was recorded as described previously (Mejías-Aponte & Kiyatkin, 2012). The animals were mounted into a stereotaxic frame (Steolting, USA) and recording electrodes were directed via a burr hole to the core portion of the VTA on the bregma zero-zero plane. The recording barrel of glass micropipette (A-M systems, USA) was filled with 2.0% of Pontamine Sky Blue solution in 0.5% sodium acetate and lowered into the brain to the VTA (Bregma=-6.84, ML=±0.5, and DV=8.6 mm from the bregma zero-zero plane). Microiontophoretic ejection was carried out by three barrel micropipettes; recording, microinjecting, and countercurrent micropipettes. Microelectrode impedance was checked (Sutter Instruments BV-10M, USA, in vitro impedance at 1000 Hz, about 5±2 MΩ) and adjusted to recording and or microiontophoretic application.
Multiunit signals were amplified (10000×) with a high impedance digital amplifier (Electromodule 3111, ScienceBeam, Tehran, Iran), filtered (bandpass 300-3000 Hz), and acquisition (sample rate=50000) by the high-speed USB port on a PC running Windows 7.0 Premium Pro. Only spontaneously active right VTA dopaminergic neurons were included in the data sheets. The electrophysiological criteria have been explained for isolation of putative VTA dopaminergic neurons in previous articles (Marinelli, Cooper, Baker, & White, 2003; Mejias-Aponte & Kiyatkin, 2012; White & Wang, 1984). Spontaneous low discharge rate (<10 spikes/second), long-duration of spikes (>2.5 ms), and a triphasic (+/-/+) or biphasic with a notch in the positive component were considered as the main criteria.
After stable firing, the baseline firing for 10 min was carried out and continued until the effect disappeared. In the control and sham groups, recordings continued about 120 min. The PSTH (peri-stimulus time histogram) of units was calculated at 1 ms bin size. On-line PSTH analysis used for detection of firing pattern change. The PSTH of all recordings was calculated by using Igor Pro 6.0 (Wavemetrics, USA) software off-line. The PCA (Principle Component Analysis) signal processing protocol with duration, amplitude, rising and falling slope criteria (±5.0% accuracy) was used for PSTH calculation. All units with no pattern changing and stable spontaneous firing were included. The obtained data in all sessions were expressed as Mean±SD.
2.4. Bupropion application
In intracerebroventricular groups, a 30-gauge stainless steel injection needle was connected to a 10-µL Hamilton syringe (Hamilton Bonaduz AG, Switzerland) with a PE-10 polyethylene tube (A-M systems, USA) used for microinjection. The injection pipette was lowered to the ipsilateral cerebral ventricle core, and the recording electrode was located into the right VTA. The drug vehicle (fresh artificial cerebrospinal fluid or ACSF) and different concentrations of bupropion were injected at 11th to 13th min with a programmable motorized microsyringe pump (New Era Pump Systems Inc., NY, USA). In the sham groups, the drug vehicle (fresh ACSF) was microinfused at the same time, and recording continued up to 120 min. In groups with intracerebroventricular microinjection, the response of 322 neurons were extracted as control (n=44), sham (n=41), and n=38, n=40, n=41, n=39, n=41, and n=38 for 1, 0.5. 0.1, 0.01, 0.001, and 0.0001 mol of bupropion, respectively.
In the second groups, microiontophoretic application of the bupropion was carried out with a microiontophoretic system (WPI 260-B, USA). Briefly, 3-barrel micropipettes (A-M Systems, USA) were pulled (David Kopf puller, 700C DKI, USA) with adequate stem and shank. One micropipette was filled with normal saline to eject a counter current, the second with recording solution as described previously, and the last one with bupropion solution (1 mol, pH=4.5). The bupropion solution was prepared by dissolving the bupropion into the sterile distilled water, and the pH of the solution was adjusted by using the 1 mol HCl and 1 mol NaOH solution. Before microiontophoretic application of the bupropion, a holding current (+20 nA) was passed through the injection micropipette, and an equal countercurrent (-20 nA) was ejected via countercurrent micropipette to cancel current effects. The ejecting currents at -50, -150, -300, and -500 nA with balanced countercurrents were used for bupropion ejection. In microiontophoretic groups, the response of 229 neurons as n=43, n=41, n=39, n=40 for -500, -300, -150, and -50 nA analysed respectively in addition to the control (n=32) and sham (n=34) groups.
2.5. Data analysis
The Kolmogorov-Smirnov test (K-S test) was used as a goodness of fit-test for the statistical probability distribution of data for parametric or non-parametric statistical tests. As mentioned previously, the single units were isolated with Igor Pro 6.0 under the PCA protocol. The PSTH was calculated off-line for all recordings. The average of the pre-microinfusion period (the first to the tenth minute) was used for each recording as a pre-stimulus period, and its firing rate was used as a baseline PSTH. The data in the post-injection period were analyzed point-to-point to determine the statistically significant difference. One-way repeated measures ANOVA with Tukey’s post hoc test was used to evaluate the statistical difference. The Igor Pro 6.0 software was used for statistical data analysis with P<0.05 as the least acceptable level of significance.
2.6. Histological verification
Pontamine Sky Blue dye was deposited at the end of the experiments for making the recording location with the passing of -20 µA electrical current through the recording electrode (~10 min). The animals were deeply anesthetized in the final session, perfused transcardially with 10% phosphate-buffered formalin solution, and their brains were removed and fixed in the perfused solution. Coronal sections (40 µm) were taken by a microtome (SLEE, London). The trajectory path and the location of the micropipette tips and infusion cannula were observed under a light microscope for verification. The mis-injected rats were excluded from the analysis.
2.7. Drugs, chemicals, and instruments
The sources of drugs, chemicals, and instruments were as follows. Bupropion, formalin, Pontamine Sky Blue, fast cresyl violet, urethane were purchased from Sigma-Aldrich (Sigma-Aldrich, USA). Sodium acetate and sodium chloride were purchased from Merck (Merck Darmstadt, Germany). Polyethylene micro-tube (A-M systems, USA) and Hamilton micro-syringes (Hamilton Bonaduz AG, Switzerland), motorized microsyringe pump (New Era Pump Systems Inc., NY, USA), microiontophoretic system (WPI 260-B, USA), micropipettes (A-M Systems, USA), pipette puller (David Kopf, 700C DKI, USA), impedance check instrument (Sutter Instruments BV-10M, USA) were prepared from standard sources.
3. Results
The data mining based on PCA and analyzing of the PSTH of neuronal firing were used for inter- and intra-group statistical analysis. The PSTH recording for 10 min before microinfusion or microiontophoresis was analyzed as a pre-stimulus time histogram. In the sham and bupropion-treated groups, the microinjection of the vehicle (fresh ACSF or quasi-saline solution, respectively) or drug solution was carried out at 11th to 13th min of the recording. Injection of drug vehicle in sham groups showed no significant difference between pre-injection and post-injection.
Briefly, the data of 485 neurons with spontaneous activity were isolated as putative VTA dopaminergic neurons and presented. Isolated 322 and 229 neurons in groups with the intracerebroventricular and microiontophoretic application of bupropion respectively were included. In the intracerebroventricular application of bupropion, 135 (42%) neurons got excited, 180 (56%) of neurons got inhibited, and 7 (2%) of them showed no response. The maximum stimulatory effects began 59.3±8.1 s post-injection on average and lasted 35.1±5.5 min, but the maximum inhibition began 46.2±7.5 s post-injection on average and lasted 42.2±08.2 min. In the microiontophoretic application of bupropion, 159 (97.5%) neurons got inhibited, and only 2 (2.5%) of them slightly got excited, but 1 neuron showed no response. The maximum inhibition lasted 51.7±18.2 min on average.
3.1. The shape of VTA dopaminergic extracellular spikes
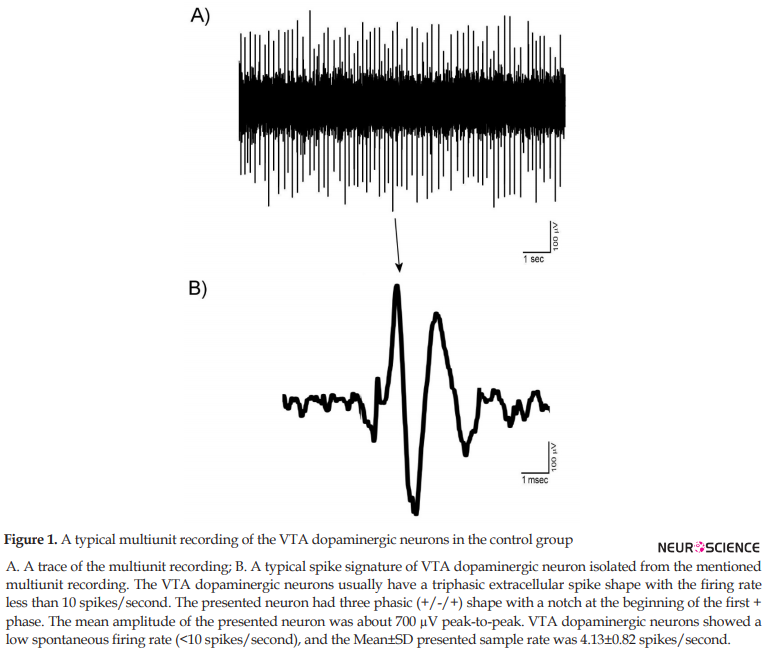
The mentioned criteria for isolation of the VTA dopaminergic neurons as the main elements of the PCA protocol used in the Igor software. The units isolated in the control groups showed the stability and reproducibility of the recordings. All isolated recordings had no sensitization-desensitization properties and pattern change. Figure 1 shows a trace (A) and a typical spike signature (B) a recording from VTA dopaminergic units. The Mean±SD peak-to-peak amplitude of the spikes was 510±46 mV. The VTA dopaminergic neurons showed a low spontaneous firing rate (<10 spikes/second).
3.2. VTA dopaminergic neuronal activity in the control and sham groups
The control group animals (naive animals) under standard condition were anesthetized by urethane and the VTA dopaminergic neurons recorded. The neuronal firing was recorded up to 2 hours for the stability of the recordings. The mean firing in each minute was calculated from the averaged firing in each second in every minute. Figure 2 shows firing of a trace (A) of a multiunit recording and the histogram (B) of the mean VTA dopaminergic neuronal firing in the control group. The mean firing rate of the selected neuron was 4.97±1.2 spikes/second during 120 min.

The sham groups in this study were intracerebroventricular sham group with microinjection of fresh ACSF and microiontophoretic application of the balanced solution at pH=4.5 that used for bupropion microiontophoretic application. Figure 3 shows the use of the fresh ACSF into the right ventricle of a sampled recording of the VTA dopaminergic neurons. The Mean±SD neuronal firing rate in the sham groups was 5.34±0.5 spikes/second. Vehicle had no significant effect on the neuronal firing. The statistical paired student t-test was used for statistical analysis.

In the microiontophoretic groups, the adjusted solution (1 mol, at pH=4.5) and ejected amounts to±500 nA of electrical current for 5 min were used in the sham group. Figure 4 shows a sample trace of the sham group VTA dopaminergic neuronal firing (A) and the histogram of the mean firing rate of the neurons in the sham group (B). The Mean±SD firing rate of the VTA dopaminergic neurons was 4.72±0.5 spikes/second. The microiontophoretic injecting of the bupropion vehicle was repeated 3 times to evaluate the precise vehicle and current effects. The ejecting current with balanced countercurrent was injected. Two-way repeated measures ANOVA was used for PSTH data, and there was no significant effect on the firing.
.PNG)
3.3. Neuronal activity of neurons following intracerebroventricular application of bupropion
The intracerebroventricular application of the bupropion showed dual, stimulatory, and inhibitory responses in the VTA dopaminergic neurons. Bupropion in the highest injected amount could excite 42% and inhibit 56% of neurons, but 2% of them showed no response. The maximum stimulatory effects began 59.3±8.1 s after intracerebroventricular infusion on average and lasted 52 min, but the maximum inhibition occurred 46.2±7.5 s after it on average and continued 78 min. The responses of the neurons were dose-dependent.
3.3.1 Stimulatory effect of the intracerebroventricular application of the bupropion
Figure 5 shows typical neuronal firing in response to the intracerebroventricular administration of the bupropion (1 mol, 5 µL/3 min, at 11th to 13th min). The PSTH of the pre-injection, post-injection, and the recovery period are also showed. In this Figure, the neuron was stimulated up to 66 min. The average±SD neuronal firing rate in the pre-injection period, was 4.8±0.24 spikes/second. The intracerebroventricular application of bupropion could excite the neuron with the Mean±SD firing rate of 7.01±1.96 spikes/second with the maximum of 10.8 spikes/second in the 33rd min after injection. In the recovery period, the neuronal firing returned to its baseline firing at 4.8±0.17 spikes/second. One-way repeated measures ANOVA with the Tukey’s post hoc test revealed that the firing rate had a significant difference (P<0.001).
.PNG)
Figure 6 shows the VTA dopaminergic neuronal firing rates of groups in response to different amounts of intracerebroventricular bupropion administration. The Figure shows that the amounts of 1, 0.5, 0.1, 0.01 mol of bupropion had significant stimulatory effects on the neurons, but 0.001 and 0.0001 mol of bupropion had no significant impact. These data revealed that intracerebroventricular application of the bupropion could excite 42% of neurons at the highest amount. The maximum firing intensity of excited neurons was 119% of the baseline firing. One-way repeated measures ANOVA with the Tukey’s post hoc test revealed that the firing rate had a significant difference (P<0.001) in the excitatory period compared with pre-injection and recovery periods. The Figure also shows that the stimulatory effect of bupropion is dose-dependent.

3.3.2 Inhibitory Effect of the intracerebroventricular application of the bupropion
Figure 7 shows the typical inhibitory response of the VTA dopaminergic neuron to the intracerebroventricular application of the bupropion (1 mol, 5 µL/3 min, at 11th to 15th min). In this Figure, the firing of the neurons decreased up to 71 min. The average±SD neuronal firing rate in the pre-injection period was 6.32±0.14 spikes/second. The intracerebroventricular microinjection of the bupropion (5 µL/3 min) inhibited the neurons. The average±SD firing of the neurons in the inhibitory period was 3.00±1.95 spikes/second with a minimum of 0 spikes/second during 44th to 48th min after the injection. In the recovery period, the neuronal firing returned to the baseline firing with 6.3±0.19 spikes/second on average. One-way repeated measures ANOVA with the Tukey’s post hoc test revealed that the firing rate had a significant difference (P<0.001).
.PNG)
Figure 8 shows the effect of intracerebroventricular microinjection of bupropion on the neuronal firing rates in the bupropion-treated groups. The Figure shows that bupropion in the amounts of 1, 0.5, 0.1, 0.01 mol had significant inhibitory effects on the neurons, but 0.001 mol of bupropion had no effect. These data revealed that intracerebroventricular microinjection of bupropion could inhibit 56% of neurons at the highest amount. The minimum firing intensity was 0% of its baseline. One-way repeated measures ANOVA with the Tukey’s post hoc test revealed that the firing rate had a significant difference (P<0.001). The Figure also shows that the stimulatory effect of bupropion is dose-dependent.

3.4. Neuronal activity of VTA dopaminergic neurons following microiontophoretic application of the bupropion
Microiontophoretic application of the bupropion in the VTA could change the dopaminergic neuronal activity. The electrical currents of -50, -150, -300, and -500 nA were applied to the bupropion solution (1 mol, pH=4.5, 5 min). In these groups, the highest ejected amount of bupropion could inhibit 97.5% of neurons, 1.8% of them had slight non-significant excitation while 0.7% (1 neuron) of neurons showed no response. In this dose, the maximum inhibition lasted 96 min with a silent period of about 30 min.
3.4.1 Inhibitory effect of microiontophoretic application of bupropion on the VTA dopaminergic neuronal activity
Microiontophoretic application of the bupropion revealed the direct effect of the bupropion on the neurons. This effect showed mainly inhibitory nature. Figure 9 shows typical neuronal inhibition by microiontophoretic application of the bupropion (-500 nA, 1 mol, pH=4.5, 5 min). The average±SD firing rate in pre-ejection period was 4.14±0.2 spikes/second. Microiontophoretic application of the bupropion inhibited the neuron with the Mean±SD firing rate of 1.5±1.4 spikes/second in the inhibitory period. The neuron was silent for 17 min during 36th to 52th min of recording. In the recovery period, the Mean±SD neuronal firing returned to baseline with 4.3±0.2 spikes/second. The neuron inhibited about 63 min on average. One-way repeated measures ANOVA with the Tukey’s post hoc test revealed that the firing rate had a significant difference (P<0.001).
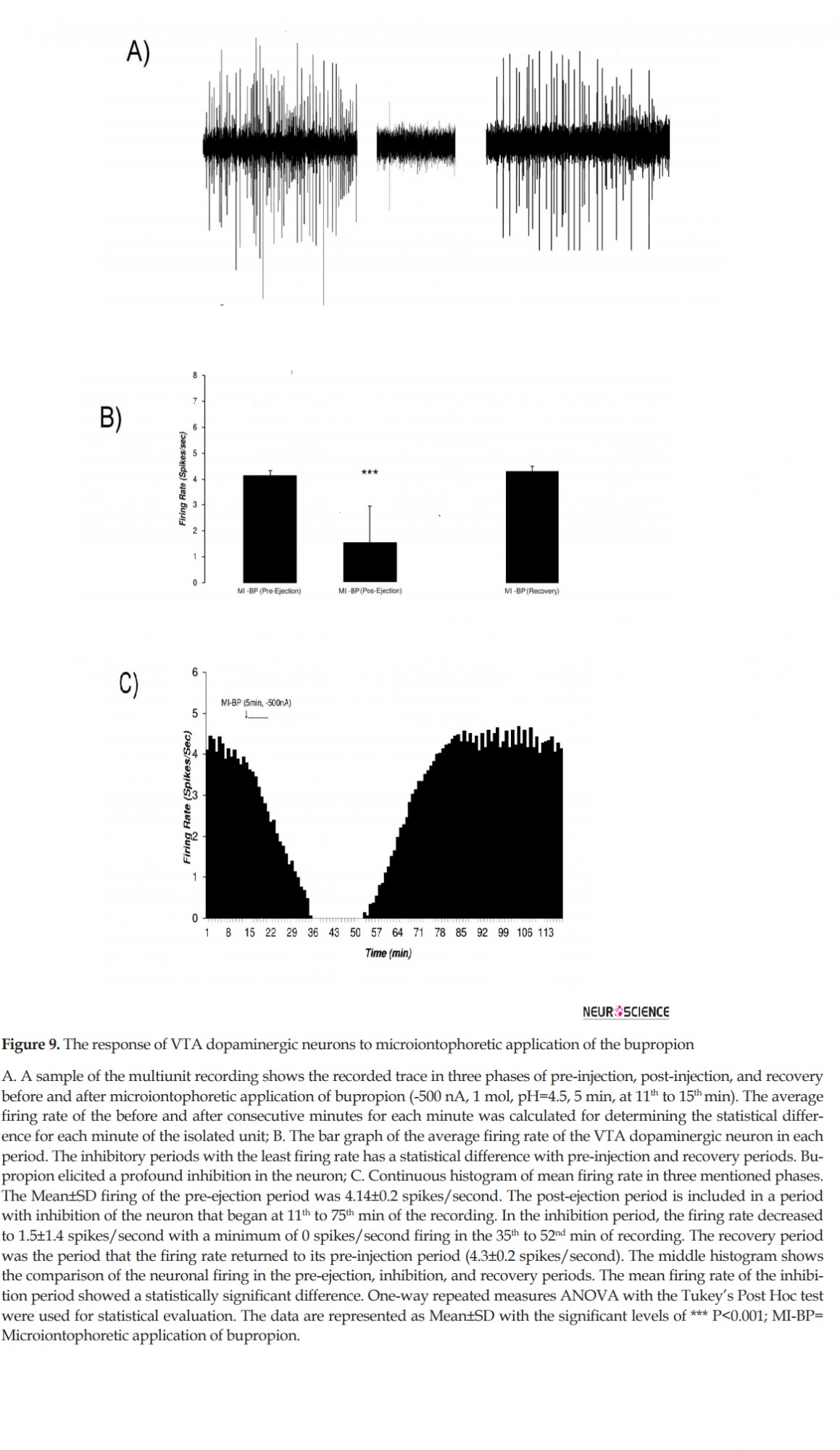
Figure 10 shows the microiontophoretic application of the bupropion with different amounts on the VTA dopaminergic neuronal firing rates. The Figure shows that the ejected amounts of -500, -300, -150 nA of bupropion could inhibit the neurons significantly. The -50 nA of bupropion had no significant effect. These data revealed that microiontophoretic application of the bupropion could inhibit 97.5% of neurons at the highest ejected amount. The minimum firing rate of the inhibited neurons was 0% of the baseline firing. The mean firing rate was 13.9%, 29.1%, 74.1% at -500, -300, and -150 nA of bupropion, respectively but at -50 nA it was 101.1% of baseline firing. One-way repeated measures ANOVA with the Tukey’s post hoc test revealed that the firing rate had a significant difference (P<0.001). The Figure also shows that the inhibitory effect of bupropion is dependent to the ejected amount.
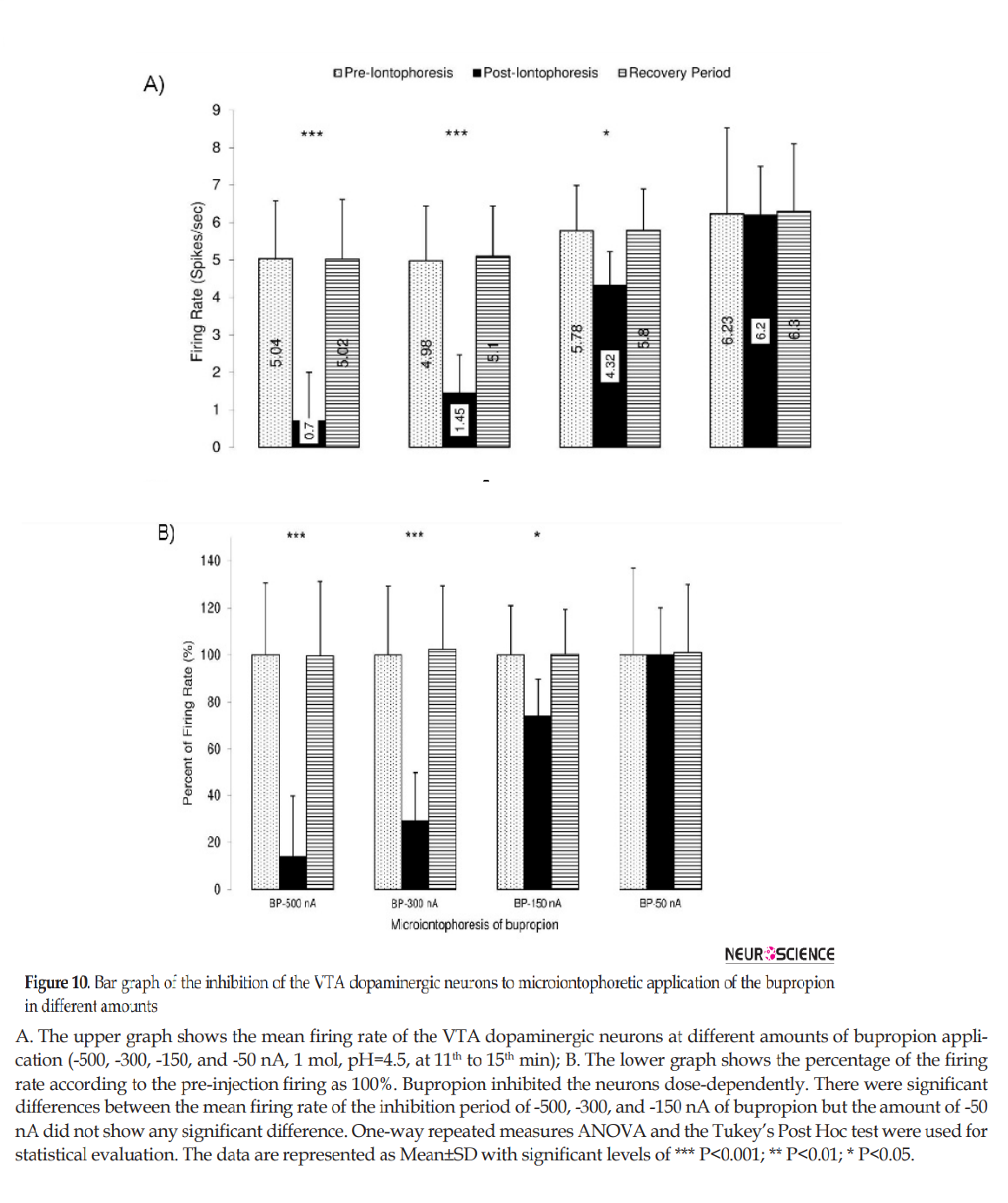
Table 1 summarizes the firing rate of the VTA dopaminergic neurons in response to intracerebroventricular and microiontophoretic administration of the bupropion. The data indicate no significant differences between the inhibitory effects of bupropion, and the direct effect of bupropion lacks stimulatory effects. Figure 11 shows the percentages of inhibited and excited neurons, inhibition and excitation duration, and the silent duration of the VTA dopaminergic neurons. The Figure shows the dose-dependent and direct inhibitory effect of bupropion.

.PNG)
As mentioned in the “Materials and Methods” section, in the final session of the experiments, the Pontamine Sky Blue was deposited in the tip location of the micropipettes by passing -20 µA of electrical currents. The brains were removed precisely after transcardial perfusion by 10% phosphate-buffered formalin solution until completion of the brain fixation. The brains were sectioned coronally to take 40-µm thick slices by a microtome. The trajectory path and location of the micropipette tips in addition to the infusion cannulae loci were evaluated. Figure 12 shows a sample of a photomicrograph on the right side of the fixed brain. In the Figure, the location of dye deposition is shown on the right side, and stereotaxic boundary of the VTA is shown on the left side.
.PNG)
4. Discussion
This study shows that acute intracerebroventricular microinjection of bupropion has dual stimulatory and inhibitory effects on putative VTA dopaminergic neurons, but the direct microiontophoretic application of the bupropion predominantly indicates inhibitory effect. The distribution of the bupropion all over the brain can affect differently VTA dopaminergic neurons directly and or indirectly. The results of this article provide the unique perspective of the direct effect of the bupropion using the microiontophoretic technique.
Depression prevalence is increasing over the past decades. Developing effective pharmaceutical agents to treat and prevent depression calls for an understanding of the etiology and mechanism of depression. The second generation of antidepressants has provided successful drugs with low side effects. The VTA and some of its targets are central to the initiation and development of drug dependence and psychiatric disorder, i.e. depression. The VTA dopaminergic neurons show different responses to antidepressant agents according to time and approach of application. This behavior reflects the existence of hidden mechanisms in the controlling of their responses. The regulation process of these neurons is complicated due to heterogeneity and differences in their pharmacological properties (Kalivas, 1993). Despite some clinical evidence suggesting the bupropion as a weak dopamine reuptake inhibitor, its mechanism of action is not well-known yet (Dong & Blier, 2001).
4.1. The VTA heterogeneity, core play roles of intra-VTA microcircuits
The different brain regions have special internal circuitry to transmit proper signals associated with their afferent nature. Immunohistochemical staining for tyrosine hydroxylase revealed diverse subcellular populations of the VTA. Dopaminergic and GABAergic populations are the first and second groups, and others are tertiary ones. This main classification has been demonstrated by electrophysiological and pharmacological evidence (Cameron, Wessendorf, & Williams, 1997). The co-release of glutamate in the axon terminals of some VTA-dopaminergic efferents in the nucleus accumbens results in further heterogeneity of the VTA (Hnasko, Hjelmstad, Fields, & Edwards, 2012; Stuber, Hnasko, Britt, Edwards, & Bonci, 2010).
Recently, the homogeneity of the VTA-dopaminergic neurons is under question due to functional differences. The most common in vivo electrophysiological studies of the VTA dopaminergic neurons propose these neurons as a single homogenous population. The simple heterogeneity of the VTA dopaminergic neurons based on electrophysiological properties is expressed on the long spike duration (>2.5 ms) and low firing rate (<10 spikes/second). The D2-agonist-induced hyperpolarization and or presence of large Ih currents due to cyclic nucleotide-dependent hyperpolarization-activated cation channels are the other tools for distinguishing putative VTA dopaminergic from non-dopaminergic neurons (Ungless & Grace, 2012). However, there are controversial findings that show the overlap of these criteria between two classes of VTA neurons (Lammel et al., 2008; Zhang, Placzek, & Dani, 2010). The diversity of putative VTA dopaminergic neurons may reflect the presence of differences in the responses of dopaminergic efferents of the VTA and the diversity of dopaminergic functional properties.
4.2. Dopamine receptors: Pre-synaptic and postsynaptic locations with D1-like and D2-like diversity
The brain dopamine receptors have different profiles concerning their location and classification. Pharmacological and genetic tools have determined multiple subtypes of the D1-like and D2-like receptors. Two major families of D1-like receptors are the D1, D5, and three major families of D2-like receptors are D2, D3, and D4 receptors. D1-like and D2-like receptors show opposite intracellular effects on the cAMP production, i.e. increasing and decreasing effects, respectively. D1-like receptors activate phospholipase C, and D2-like receptors activate K channels and inhibit calcium channels (Spano, Govoni, & Trabucchi, 1978). The modulatory effects of the dopamine receptors on the neuron can produce a cascade of protein kinase A and after-activation of terminal signaling proteins (Beaulieu & Gainetdinov, 2011). The density of the D1-like and D2-like dopamine receptors are different in the brain regions. D1-like receptors are the dominant form of dopamine receptors in the postsynaptic non-dopaminergic neurons. High expression of D1-like receptors was found in the frontal cortex, substantia nigra, striatum, olfactory bulb, and amygdala. D2-like receptors are predominantly found in the dopaminergic as well as in non-dopaminergic neurons postsynaptically. However, in dopaminergic neurons, the massive D2-like receptors are found in the pre-synaptic membrane and somatodendritic loci. D2-receptors with average densities are located in the substantia nigra, ventral tegmental area, hypothalamus, septum, amygdala, hippocampus, and the cortical regions (Beaulieu & Gainetdinov, 2011; Moritz, Free, & Sibley, 2018).
Reuptake inhibition of the multi-neurotransmitters has provided a novel and effective generation of antidepressants, such as bupropion (Dremencov et al., 2004; Dremencov et al., 2005; Nutt et al., 2006). Bupropion (Wellbutrin) that is recently re-marketed in the USA and other regions exerts its antidepressant effect by blocking the reuptake of dopamine. Bupropion has a weak inhibitory effect on norepinephrine reuptake, too (Horst & Preskorn, 1998). The confirmed anti-smoke effects of bupropion are believed to antagonize the nicotinic acetylcholine receptors (nAchRs) (Taeron, 2002). Although it is reported that bupropion, primarily blocks the reuptake of both dopamine and norepinephrine, the mechanism of its action has not been fully elucidated (Ascher et al., 1995). The dual effect of bupropion on the dopamine and norepinephrine reuptakes has changed the treatment of specific symptoms of depression, cognitive, and fatigue associated with selective monoamine inhibitors and selective serotonin reuptake inhibitors (SSRIs) (Fabre, Brodie, Garver, & Zung,1983; Green, 1997). Bupropion also has less risk of sexual dysfunction compared to SSRIs (Pereira, Arias-Carrion, Machado, Nardi, & Silva, 2014).
Although bupropion primarily inhibits the dopamine reuptake, its precise mechanism of action is still unknown because of differences in its in vivo and in vitro effects. The in vitro potency of norepinephrine reuptake inhibition of bupropion is about twice than that in vivo (Ascher et al., 1995).
The duration of its administration has different effects on norepinephrine-releasing neurons. In acute systemic application, it can reduce norepinephrine releasing firing rates, but its sustained administration can recover the baseline firing rate (Ghanbari, El Mansari, & Blier, 2010). This effect is suggested due to the somatodendritic α2-adrenoceptors activation (Dong & Blier, 2001). Besides its action on monoamines, a low amount of bupropion can attenuate nicotine-evoked dopamine release in striatum in slice preparations (Mansvelder, Fagen, Chang, Mitchum, & McGehee, 2007; Sidhpura, Redfern, Rowley, Heal, & Wonnacott, 2007). Nicotine can induce significantly large somatic responses in the neurons (Klink, de Kerchove d’Exaerde, Zoli, & Changeux, 2001; Wooltorton, Pidoplichko, Broide, & Dani, 2003) via alpha7-nicotinic receptors and modulate nicotine-induced reinforcement and extracellular dopamine in the mesolimbic system (Besson et al., 2012). The mechanism of the bupropion action on the VTA dopaminergic neurons is not well-known due to the presence of different dopamine receptors and their various locations and destinations pre- and postsynaptically.
Our recent studies showed that acute systemic infusion of bupropion could decrease formalin-induced pain behavior in rats (Naderi, Pakdel, Osalou, & Cankurt, 2014). Microinfusion of the bupropion into the locus coeruleus nuclease could decrease formalin-induced pain response (Jahanbani et al., 2014). The intracerebroventricular application of the bupropion revealed that it could reduce the spontaneous firing of locus coeruleus neurons dose-dependently (Pakdel et al., 2014). The effect of the bupropion on the dendritic adrenergic receptors has been postulated as a critical factor for these actions. It is reported that neither 2 nor 14 days of bupropion (30 mg/kg/d) administration alter the firing and burst activity of dopamine neurons (El Mansari, Ghanbari, Janssen, & Blier, 2008), but there are other reports about increase in firing rate with 14 days administration of bupropion at two 10 and 20 mg/kg/d doses (West & Weiss, 2011). Cooper et al. revealed that the acute systemic infusion of bupropion could inhibit the firing of the VTA dopaminergic neurons (Cooper et al., 1994).
Bupropion can also act on the VTA dopaminergic neurons by VTA GABAergic neurons. The microinfusion of the bupropion could inhibit the neuronal firing of the putative VTA GABAergic neurons dose-dependently (Amirabadi et al., 2014). We hypothesized that the critical neuronal control of the VTA dopaminergic neurons is the GABA neurotransmission and because there is no evidence about the direct effect of bupropion on VTA dopaminergic neurons, this study was designed to test the acute effects of bupropion via intracerebroventricular and microiontophoretic applications on the dopaminergic neuronal firing rates of the VTA.
2. Methods
2.1. Ethical approval
The committee for biomedical research ethics approval reviewed the procedure, experiments, protocols, and all guidelines for the care and use of experimental animals. The Animal Laboratory Center of Urmia University of Medical Sciences (ALCUUMS) supervised precisely over all ethical issues. All experimental procedures and protocols were approved by the Urmia Medical Science Research Ethics Committee (UMSREC). The ethical issues were performed in accordance with the National Institute of Health (NIH) guide for the care and use of laboratory animals.
2.2. Study animals
Healthy male Wistar rats of 250-280 g weight (Pasteur Institute, Tehran, Iran) were housed (three on a cage) at a 12/12 h light/dark cycle (7:00 AM to 7:00 PM). The ambient temperature was controlled (22±2°C), and the food and water were at labium. The two intracerebroventricular and microiontophoretic categories of animals were divided into 14 groups (5 per each subgroup) according to the bupropion application with the sham and control groups too. Apart from the control and sham groups, in the first category, the animals received 1, 0.5, 0.1, 0.01, 0.001, and 0.0001 mol of intracerebroventricular bupropion (5 μL/3 min). In the second category, bupropion was ejected by -50, -150, -300, and -500 nA electrical currents (1 mol of bupropion solution, pH=4.5, 5min) with the control and sham subgroups. Briefly, animals were anesthetized by urethane (1.2 g/kg) initially, and the booster dose (~ 15% to 25% of the initial dose) was used when there were any discomfort signs. The core body temperature was continuously monitored and maintained at ~37°C during the entire experiment. The stereotaxic coordinations of bregma and interaural landmarks were determined for each rat, and the location of recording and injecting electrodes were exposed according to the Paxinos and Watson stereotaxic rat brain atlas (Paxinos & Watson, 2007).
2.3. VTA multiple electrophysiological recording
Multiunit extracellular electrophysiological activity of the VTA dopaminergic neurons was recorded as described previously (Mejías-Aponte & Kiyatkin, 2012). The animals were mounted into a stereotaxic frame (Steolting, USA) and recording electrodes were directed via a burr hole to the core portion of the VTA on the bregma zero-zero plane. The recording barrel of glass micropipette (A-M systems, USA) was filled with 2.0% of Pontamine Sky Blue solution in 0.5% sodium acetate and lowered into the brain to the VTA (Bregma=-6.84, ML=±0.5, and DV=8.6 mm from the bregma zero-zero plane). Microiontophoretic ejection was carried out by three barrel micropipettes; recording, microinjecting, and countercurrent micropipettes. Microelectrode impedance was checked (Sutter Instruments BV-10M, USA, in vitro impedance at 1000 Hz, about 5±2 MΩ) and adjusted to recording and or microiontophoretic application.
Multiunit signals were amplified (10000×) with a high impedance digital amplifier (Electromodule 3111, ScienceBeam, Tehran, Iran), filtered (bandpass 300-3000 Hz), and acquisition (sample rate=50000) by the high-speed USB port on a PC running Windows 7.0 Premium Pro. Only spontaneously active right VTA dopaminergic neurons were included in the data sheets. The electrophysiological criteria have been explained for isolation of putative VTA dopaminergic neurons in previous articles (Marinelli, Cooper, Baker, & White, 2003; Mejias-Aponte & Kiyatkin, 2012; White & Wang, 1984). Spontaneous low discharge rate (<10 spikes/second), long-duration of spikes (>2.5 ms), and a triphasic (+/-/+) or biphasic with a notch in the positive component were considered as the main criteria.
After stable firing, the baseline firing for 10 min was carried out and continued until the effect disappeared. In the control and sham groups, recordings continued about 120 min. The PSTH (peri-stimulus time histogram) of units was calculated at 1 ms bin size. On-line PSTH analysis used for detection of firing pattern change. The PSTH of all recordings was calculated by using Igor Pro 6.0 (Wavemetrics, USA) software off-line. The PCA (Principle Component Analysis) signal processing protocol with duration, amplitude, rising and falling slope criteria (±5.0% accuracy) was used for PSTH calculation. All units with no pattern changing and stable spontaneous firing were included. The obtained data in all sessions were expressed as Mean±SD.
2.4. Bupropion application
In intracerebroventricular groups, a 30-gauge stainless steel injection needle was connected to a 10-µL Hamilton syringe (Hamilton Bonaduz AG, Switzerland) with a PE-10 polyethylene tube (A-M systems, USA) used for microinjection. The injection pipette was lowered to the ipsilateral cerebral ventricle core, and the recording electrode was located into the right VTA. The drug vehicle (fresh artificial cerebrospinal fluid or ACSF) and different concentrations of bupropion were injected at 11th to 13th min with a programmable motorized microsyringe pump (New Era Pump Systems Inc., NY, USA). In the sham groups, the drug vehicle (fresh ACSF) was microinfused at the same time, and recording continued up to 120 min. In groups with intracerebroventricular microinjection, the response of 322 neurons were extracted as control (n=44), sham (n=41), and n=38, n=40, n=41, n=39, n=41, and n=38 for 1, 0.5. 0.1, 0.01, 0.001, and 0.0001 mol of bupropion, respectively.
In the second groups, microiontophoretic application of the bupropion was carried out with a microiontophoretic system (WPI 260-B, USA). Briefly, 3-barrel micropipettes (A-M Systems, USA) were pulled (David Kopf puller, 700C DKI, USA) with adequate stem and shank. One micropipette was filled with normal saline to eject a counter current, the second with recording solution as described previously, and the last one with bupropion solution (1 mol, pH=4.5). The bupropion solution was prepared by dissolving the bupropion into the sterile distilled water, and the pH of the solution was adjusted by using the 1 mol HCl and 1 mol NaOH solution. Before microiontophoretic application of the bupropion, a holding current (+20 nA) was passed through the injection micropipette, and an equal countercurrent (-20 nA) was ejected via countercurrent micropipette to cancel current effects. The ejecting currents at -50, -150, -300, and -500 nA with balanced countercurrents were used for bupropion ejection. In microiontophoretic groups, the response of 229 neurons as n=43, n=41, n=39, n=40 for -500, -300, -150, and -50 nA analysed respectively in addition to the control (n=32) and sham (n=34) groups.
2.5. Data analysis
The Kolmogorov-Smirnov test (K-S test) was used as a goodness of fit-test for the statistical probability distribution of data for parametric or non-parametric statistical tests. As mentioned previously, the single units were isolated with Igor Pro 6.0 under the PCA protocol. The PSTH was calculated off-line for all recordings. The average of the pre-microinfusion period (the first to the tenth minute) was used for each recording as a pre-stimulus period, and its firing rate was used as a baseline PSTH. The data in the post-injection period were analyzed point-to-point to determine the statistically significant difference. One-way repeated measures ANOVA with Tukey’s post hoc test was used to evaluate the statistical difference. The Igor Pro 6.0 software was used for statistical data analysis with P<0.05 as the least acceptable level of significance.
2.6. Histological verification
Pontamine Sky Blue dye was deposited at the end of the experiments for making the recording location with the passing of -20 µA electrical current through the recording electrode (~10 min). The animals were deeply anesthetized in the final session, perfused transcardially with 10% phosphate-buffered formalin solution, and their brains were removed and fixed in the perfused solution. Coronal sections (40 µm) were taken by a microtome (SLEE, London). The trajectory path and the location of the micropipette tips and infusion cannula were observed under a light microscope for verification. The mis-injected rats were excluded from the analysis.
2.7. Drugs, chemicals, and instruments
The sources of drugs, chemicals, and instruments were as follows. Bupropion, formalin, Pontamine Sky Blue, fast cresyl violet, urethane were purchased from Sigma-Aldrich (Sigma-Aldrich, USA). Sodium acetate and sodium chloride were purchased from Merck (Merck Darmstadt, Germany). Polyethylene micro-tube (A-M systems, USA) and Hamilton micro-syringes (Hamilton Bonaduz AG, Switzerland), motorized microsyringe pump (New Era Pump Systems Inc., NY, USA), microiontophoretic system (WPI 260-B, USA), micropipettes (A-M Systems, USA), pipette puller (David Kopf, 700C DKI, USA), impedance check instrument (Sutter Instruments BV-10M, USA) were prepared from standard sources.
3. Results
The data mining based on PCA and analyzing of the PSTH of neuronal firing were used for inter- and intra-group statistical analysis. The PSTH recording for 10 min before microinfusion or microiontophoresis was analyzed as a pre-stimulus time histogram. In the sham and bupropion-treated groups, the microinjection of the vehicle (fresh ACSF or quasi-saline solution, respectively) or drug solution was carried out at 11th to 13th min of the recording. Injection of drug vehicle in sham groups showed no significant difference between pre-injection and post-injection.
Briefly, the data of 485 neurons with spontaneous activity were isolated as putative VTA dopaminergic neurons and presented. Isolated 322 and 229 neurons in groups with the intracerebroventricular and microiontophoretic application of bupropion respectively were included. In the intracerebroventricular application of bupropion, 135 (42%) neurons got excited, 180 (56%) of neurons got inhibited, and 7 (2%) of them showed no response. The maximum stimulatory effects began 59.3±8.1 s post-injection on average and lasted 35.1±5.5 min, but the maximum inhibition began 46.2±7.5 s post-injection on average and lasted 42.2±08.2 min. In the microiontophoretic application of bupropion, 159 (97.5%) neurons got inhibited, and only 2 (2.5%) of them slightly got excited, but 1 neuron showed no response. The maximum inhibition lasted 51.7±18.2 min on average.
3.1. The shape of VTA dopaminergic extracellular spikes
The mentioned criteria for isolation of the VTA dopaminergic neurons as the main elements of the PCA protocol used in the Igor software. The units isolated in the control groups showed the stability and reproducibility of the recordings. All isolated recordings had no sensitization-desensitization properties and pattern change. Figure 1 shows a trace (A) and a typical spike signature (B) a recording from VTA dopaminergic units. The Mean±SD peak-to-peak amplitude of the spikes was 510±46 mV. The VTA dopaminergic neurons showed a low spontaneous firing rate (<10 spikes/second).
3.2. VTA dopaminergic neuronal activity in the control and sham groups
The control group animals (naive animals) under standard condition were anesthetized by urethane and the VTA dopaminergic neurons recorded. The neuronal firing was recorded up to 2 hours for the stability of the recordings. The mean firing in each minute was calculated from the averaged firing in each second in every minute. Figure 2 shows firing of a trace (A) of a multiunit recording and the histogram (B) of the mean VTA dopaminergic neuronal firing in the control group. The mean firing rate of the selected neuron was 4.97±1.2 spikes/second during 120 min.
The sham groups in this study were intracerebroventricular sham group with microinjection of fresh ACSF and microiontophoretic application of the balanced solution at pH=4.5 that used for bupropion microiontophoretic application. Figure 3 shows the use of the fresh ACSF into the right ventricle of a sampled recording of the VTA dopaminergic neurons. The Mean±SD neuronal firing rate in the sham groups was 5.34±0.5 spikes/second. Vehicle had no significant effect on the neuronal firing. The statistical paired student t-test was used for statistical analysis.
In the microiontophoretic groups, the adjusted solution (1 mol, at pH=4.5) and ejected amounts to±500 nA of electrical current for 5 min were used in the sham group. Figure 4 shows a sample trace of the sham group VTA dopaminergic neuronal firing (A) and the histogram of the mean firing rate of the neurons in the sham group (B). The Mean±SD firing rate of the VTA dopaminergic neurons was 4.72±0.5 spikes/second. The microiontophoretic injecting of the bupropion vehicle was repeated 3 times to evaluate the precise vehicle and current effects. The ejecting current with balanced countercurrent was injected. Two-way repeated measures ANOVA was used for PSTH data, and there was no significant effect on the firing.
.PNG)
3.3. Neuronal activity of neurons following intracerebroventricular application of bupropion
The intracerebroventricular application of the bupropion showed dual, stimulatory, and inhibitory responses in the VTA dopaminergic neurons. Bupropion in the highest injected amount could excite 42% and inhibit 56% of neurons, but 2% of them showed no response. The maximum stimulatory effects began 59.3±8.1 s after intracerebroventricular infusion on average and lasted 52 min, but the maximum inhibition occurred 46.2±7.5 s after it on average and continued 78 min. The responses of the neurons were dose-dependent.
3.3.1 Stimulatory effect of the intracerebroventricular application of the bupropion
Figure 5 shows typical neuronal firing in response to the intracerebroventricular administration of the bupropion (1 mol, 5 µL/3 min, at 11th to 13th min). The PSTH of the pre-injection, post-injection, and the recovery period are also showed. In this Figure, the neuron was stimulated up to 66 min. The average±SD neuronal firing rate in the pre-injection period, was 4.8±0.24 spikes/second. The intracerebroventricular application of bupropion could excite the neuron with the Mean±SD firing rate of 7.01±1.96 spikes/second with the maximum of 10.8 spikes/second in the 33rd min after injection. In the recovery period, the neuronal firing returned to its baseline firing at 4.8±0.17 spikes/second. One-way repeated measures ANOVA with the Tukey’s post hoc test revealed that the firing rate had a significant difference (P<0.001).
Figure 6 shows the VTA dopaminergic neuronal firing rates of groups in response to different amounts of intracerebroventricular bupropion administration. The Figure shows that the amounts of 1, 0.5, 0.1, 0.01 mol of bupropion had significant stimulatory effects on the neurons, but 0.001 and 0.0001 mol of bupropion had no significant impact. These data revealed that intracerebroventricular application of the bupropion could excite 42% of neurons at the highest amount. The maximum firing intensity of excited neurons was 119% of the baseline firing. One-way repeated measures ANOVA with the Tukey’s post hoc test revealed that the firing rate had a significant difference (P<0.001) in the excitatory period compared with pre-injection and recovery periods. The Figure also shows that the stimulatory effect of bupropion is dose-dependent.
3.3.2 Inhibitory Effect of the intracerebroventricular application of the bupropion
Figure 7 shows the typical inhibitory response of the VTA dopaminergic neuron to the intracerebroventricular application of the bupropion (1 mol, 5 µL/3 min, at 11th to 15th min). In this Figure, the firing of the neurons decreased up to 71 min. The average±SD neuronal firing rate in the pre-injection period was 6.32±0.14 spikes/second. The intracerebroventricular microinjection of the bupropion (5 µL/3 min) inhibited the neurons. The average±SD firing of the neurons in the inhibitory period was 3.00±1.95 spikes/second with a minimum of 0 spikes/second during 44th to 48th min after the injection. In the recovery period, the neuronal firing returned to the baseline firing with 6.3±0.19 spikes/second on average. One-way repeated measures ANOVA with the Tukey’s post hoc test revealed that the firing rate had a significant difference (P<0.001).
Figure 8 shows the effect of intracerebroventricular microinjection of bupropion on the neuronal firing rates in the bupropion-treated groups. The Figure shows that bupropion in the amounts of 1, 0.5, 0.1, 0.01 mol had significant inhibitory effects on the neurons, but 0.001 mol of bupropion had no effect. These data revealed that intracerebroventricular microinjection of bupropion could inhibit 56% of neurons at the highest amount. The minimum firing intensity was 0% of its baseline. One-way repeated measures ANOVA with the Tukey’s post hoc test revealed that the firing rate had a significant difference (P<0.001). The Figure also shows that the stimulatory effect of bupropion is dose-dependent.
3.4. Neuronal activity of VTA dopaminergic neurons following microiontophoretic application of the bupropion
Microiontophoretic application of the bupropion in the VTA could change the dopaminergic neuronal activity. The electrical currents of -50, -150, -300, and -500 nA were applied to the bupropion solution (1 mol, pH=4.5, 5 min). In these groups, the highest ejected amount of bupropion could inhibit 97.5% of neurons, 1.8% of them had slight non-significant excitation while 0.7% (1 neuron) of neurons showed no response. In this dose, the maximum inhibition lasted 96 min with a silent period of about 30 min.
3.4.1 Inhibitory effect of microiontophoretic application of bupropion on the VTA dopaminergic neuronal activity
Microiontophoretic application of the bupropion revealed the direct effect of the bupropion on the neurons. This effect showed mainly inhibitory nature. Figure 9 shows typical neuronal inhibition by microiontophoretic application of the bupropion (-500 nA, 1 mol, pH=4.5, 5 min). The average±SD firing rate in pre-ejection period was 4.14±0.2 spikes/second. Microiontophoretic application of the bupropion inhibited the neuron with the Mean±SD firing rate of 1.5±1.4 spikes/second in the inhibitory period. The neuron was silent for 17 min during 36th to 52th min of recording. In the recovery period, the Mean±SD neuronal firing returned to baseline with 4.3±0.2 spikes/second. The neuron inhibited about 63 min on average. One-way repeated measures ANOVA with the Tukey’s post hoc test revealed that the firing rate had a significant difference (P<0.001).
Figure 10 shows the microiontophoretic application of the bupropion with different amounts on the VTA dopaminergic neuronal firing rates. The Figure shows that the ejected amounts of -500, -300, -150 nA of bupropion could inhibit the neurons significantly. The -50 nA of bupropion had no significant effect. These data revealed that microiontophoretic application of the bupropion could inhibit 97.5% of neurons at the highest ejected amount. The minimum firing rate of the inhibited neurons was 0% of the baseline firing. The mean firing rate was 13.9%, 29.1%, 74.1% at -500, -300, and -150 nA of bupropion, respectively but at -50 nA it was 101.1% of baseline firing. One-way repeated measures ANOVA with the Tukey’s post hoc test revealed that the firing rate had a significant difference (P<0.001). The Figure also shows that the inhibitory effect of bupropion is dependent to the ejected amount.
Table 1 summarizes the firing rate of the VTA dopaminergic neurons in response to intracerebroventricular and microiontophoretic administration of the bupropion. The data indicate no significant differences between the inhibitory effects of bupropion, and the direct effect of bupropion lacks stimulatory effects. Figure 11 shows the percentages of inhibited and excited neurons, inhibition and excitation duration, and the silent duration of the VTA dopaminergic neurons. The Figure shows the dose-dependent and direct inhibitory effect of bupropion.
As mentioned in the “Materials and Methods” section, in the final session of the experiments, the Pontamine Sky Blue was deposited in the tip location of the micropipettes by passing -20 µA of electrical currents. The brains were removed precisely after transcardial perfusion by 10% phosphate-buffered formalin solution until completion of the brain fixation. The brains were sectioned coronally to take 40-µm thick slices by a microtome. The trajectory path and location of the micropipette tips in addition to the infusion cannulae loci were evaluated. Figure 12 shows a sample of a photomicrograph on the right side of the fixed brain. In the Figure, the location of dye deposition is shown on the right side, and stereotaxic boundary of the VTA is shown on the left side.
4. Discussion
This study shows that acute intracerebroventricular microinjection of bupropion has dual stimulatory and inhibitory effects on putative VTA dopaminergic neurons, but the direct microiontophoretic application of the bupropion predominantly indicates inhibitory effect. The distribution of the bupropion all over the brain can affect differently VTA dopaminergic neurons directly and or indirectly. The results of this article provide the unique perspective of the direct effect of the bupropion using the microiontophoretic technique.
Depression prevalence is increasing over the past decades. Developing effective pharmaceutical agents to treat and prevent depression calls for an understanding of the etiology and mechanism of depression. The second generation of antidepressants has provided successful drugs with low side effects. The VTA and some of its targets are central to the initiation and development of drug dependence and psychiatric disorder, i.e. depression. The VTA dopaminergic neurons show different responses to antidepressant agents according to time and approach of application. This behavior reflects the existence of hidden mechanisms in the controlling of their responses. The regulation process of these neurons is complicated due to heterogeneity and differences in their pharmacological properties (Kalivas, 1993). Despite some clinical evidence suggesting the bupropion as a weak dopamine reuptake inhibitor, its mechanism of action is not well-known yet (Dong & Blier, 2001).
4.1. The VTA heterogeneity, core play roles of intra-VTA microcircuits
The different brain regions have special internal circuitry to transmit proper signals associated with their afferent nature. Immunohistochemical staining for tyrosine hydroxylase revealed diverse subcellular populations of the VTA. Dopaminergic and GABAergic populations are the first and second groups, and others are tertiary ones. This main classification has been demonstrated by electrophysiological and pharmacological evidence (Cameron, Wessendorf, & Williams, 1997). The co-release of glutamate in the axon terminals of some VTA-dopaminergic efferents in the nucleus accumbens results in further heterogeneity of the VTA (Hnasko, Hjelmstad, Fields, & Edwards, 2012; Stuber, Hnasko, Britt, Edwards, & Bonci, 2010).
Recently, the homogeneity of the VTA-dopaminergic neurons is under question due to functional differences. The most common in vivo electrophysiological studies of the VTA dopaminergic neurons propose these neurons as a single homogenous population. The simple heterogeneity of the VTA dopaminergic neurons based on electrophysiological properties is expressed on the long spike duration (>2.5 ms) and low firing rate (<10 spikes/second). The D2-agonist-induced hyperpolarization and or presence of large Ih currents due to cyclic nucleotide-dependent hyperpolarization-activated cation channels are the other tools for distinguishing putative VTA dopaminergic from non-dopaminergic neurons (Ungless & Grace, 2012). However, there are controversial findings that show the overlap of these criteria between two classes of VTA neurons (Lammel et al., 2008; Zhang, Placzek, & Dani, 2010). The diversity of putative VTA dopaminergic neurons may reflect the presence of differences in the responses of dopaminergic efferents of the VTA and the diversity of dopaminergic functional properties.
4.2. Dopamine receptors: Pre-synaptic and postsynaptic locations with D1-like and D2-like diversity
The brain dopamine receptors have different profiles concerning their location and classification. Pharmacological and genetic tools have determined multiple subtypes of the D1-like and D2-like receptors. Two major families of D1-like receptors are the D1, D5, and three major families of D2-like receptors are D2, D3, and D4 receptors. D1-like and D2-like receptors show opposite intracellular effects on the cAMP production, i.e. increasing and decreasing effects, respectively. D1-like receptors activate phospholipase C, and D2-like receptors activate K channels and inhibit calcium channels (Spano, Govoni, & Trabucchi, 1978). The modulatory effects of the dopamine receptors on the neuron can produce a cascade of protein kinase A and after-activation of terminal signaling proteins (Beaulieu & Gainetdinov, 2011). The density of the D1-like and D2-like dopamine receptors are different in the brain regions. D1-like receptors are the dominant form of dopamine receptors in the postsynaptic non-dopaminergic neurons. High expression of D1-like receptors was found in the frontal cortex, substantia nigra, striatum, olfactory bulb, and amygdala. D2-like receptors are predominantly found in the dopaminergic as well as in non-dopaminergic neurons postsynaptically. However, in dopaminergic neurons, the massive D2-like receptors are found in the pre-synaptic membrane and somatodendritic loci. D2-receptors with average densities are located in the substantia nigra, ventral tegmental area, hypothalamus, septum, amygdala, hippocampus, and the cortical regions (Beaulieu & Gainetdinov, 2011; Moritz, Free, & Sibley, 2018).
D1-like dopamine receptors are rare in the VTA dopaminergic neurons, and D2-like receptors have important roles. The presence of the D1-like receptors in the VTA non-dopaminergic neurons is predominate, and their agonist can create the stimulatory effect on VTA dopaminergic neurons indirectly.
4.3. Stimulatory effects of bupropion on the VTA dopaminergic neurons: The prominent indirect effect
The stimulatory effects of an antidepressant may arise from the monoamine hypothesis of depression and its pharmacotherapy. The first idea about the mechanism of the stimulatory effects of bupropion on the VTA dopaminergic neurons is based on the overall impact of antidepressants. The stimulatory effects of bupropion may reflect the presence of the stimulatory receptors on the VTA dopaminergic neurons for dopamine because bupropion inhibition of dopamine reuptake can increase dopamine availability to produce excitation. The direct dopamine stimulatory effects on the VTA dopaminergic neurons may be produced by D1-like receptors on the VTA dopaminergic neurons while there is no substantial evidence to support this hypothesis. The density of D1-like receptors on the VTA dopaminergic neurons is very low compared with the density of D2-like receptors.
Direct stimulatory effects of bupropion on the VTA dopaminergic neurons can be due to the action of the dopamine transporter to induce excitation because of its coupling to chloride ions (Horn, 1990). This activity, as seen in the amphetamine fast ionotropic excitatory response to depolarization, is caused by the inhibition of the chloride influx. The fast in vitro amphetamine-induced currents is comparable with glutamatergic ionotropic currents (DeFelice & Goswami, 2007).
The indirect stimulation of the VTA dopaminergic neurons is the most acceptable hypothesis. The GABAergic input to the VTA dopaminergic neurons is one of the major inhibitory inputs, and bupropion alone can reduce the inhibitory GABAergic input to dopaminergic neurons. The VTA dopaminergic neurons are under the tonic discharge of GABAergic inputs from medium spiny neurons of the nucleus accumbens. These neurons exert a tonic inhibitory effect on the VTA dopaminergic neurons and suppress their activity (Haber, Wolfe, & Groenewegen, 1990; Kalivas, Churchill, & Klitenick, 1993; Nauta, Smith, Faull, & Domesick, 1978). The nucleus accumbens has a noticeable effect on the neuronal activity of VTA and VTA-nucleus accumbens interaction has a key role in the firing rate (Moaddab, Kermani, Azizi, & Haghparast, 2013).
On the other hand, bupropion as a nicotinic acetylcholine receptors (nAChRs) antagonist can inhibit nicotine effects on GABA neurons, and the nAChRs blockade can remove the excitatory drive or disinhibition to GABA neurons (Fryer & Lukas, 1999; Slemmer, Martin, & Damaj, 2000). The GABAergic innervation blockade enhances neuronal activity with a disinhibition mechanism (Johnson & North, 1992). The nAChRs in the VTA neurons can also contribute to their nicotine rewarding effects (Nisell, Nomikos, & Svensson, 1994).
The excitation of VTA dopaminergic neurons by acute micro-infusion of bupropion is consistent with the emerging views on the dopamine role in reward-based learning and memory processes (Berridge, 2007; De Biasi & Dani, 2011; Schultz, 2007). Increase in neuronal activity of VTA can amplify and synchronize the output of DA signaling to different targets such as the nucleus accumbens. Recent studies show that VTA neuronal activity enhances rewards and correlates with the increase in synchronization of neuronal spiking (Cryan, Bruijnzeel, Skjei, & Markou, 2003; De Biasi & Dani, 2011; Joshua et al., 2009).
Inhibition of the dopamine reuptake can increase the synaptic dopamine content. The onset of bupropion excitatory effect on the neurons has many delays in comparison with its inhibitory effects. This difference indicates that excitation could be generated by afferent neurons, not by their somatodendritic receptors. Increase in discharge rate by bupropion can also be explained by a decrease in tonic inhibitory inputs to the VTA. In this study, the changes in neuronal activity, excitation, and inhibition began 60±10.5 s and 8±4.4 s respectively after the maximum amount of bupropion. This delay in the onset of bupropion effects suggests that its excitatory effects on the neurons are carried out via indirect effect or probable disinhibition of GABAergic inputs to the VTA. We propose that different time courses from inactivation of nAChR subtypes on GABA inputs to the neurons can contribute to the bupropion-delayed excitatory effect. The distinct contributions of different nAChRs in VTA dopaminergic neurons have been already defined in many studies. In addition to nicotine, activation of α4α6β2* nAChRs in the neurons is sufficient to support the initiation of cellular changes. α4α6β2* nAChRs may be a promising target for future drug addiction (Engle, Shih, McIntosh, & Drenan, 2013). The postulated differences in distinct contributions of α4* and α6* nAChRs in the soma vs. the axon terminal of nucleus accumbens neurons are related to neuronal firing properties (Exley et al., 2011).
4.4. Inhibitory effects of bupropion on the VTA dopaminergic neurons: The prominent direct effect
Based on our study results, the inhibitory effects of the microiontophoretic application of bupropion were much stronger than its excitatory effects on VTA dopaminergic neuronal activity. Several possible mechanisms can cause inhibitory effects.
The direct inhibitory effects of the bupropion on VTA dopaminergic neurons are due to the occupation of the D2-like dopamine receptors. The major D2-like dopamine receptors are autoreceptors on the somatodendritic regions and strongly inhibit the VTA-dopaminergic neurons by lowering intracellular cAMP. Lejeune and Millan (1995) reported that the systemic application of the D3 dopamine receptor selective agonists could inhibit VTA dopaminergic neuronal firing rate that is blocked by D3 dopamine receptor antagonists’ dose-dependently both of them (Lejeune & Millan, 1995). The activation of the D2 dopamine receptors on the VTA dopaminergic neurons in the wild-type mice could produce profound hyperpolarization, but in the D2-/- mice, it was abolished due to receptor lacking. However, the subtype of the D2 dopamine receptor is essential because of its response to activation. On the other hand, the overall results show that the D2 dopamine autoreceptors on the VTA dopaminergic neurons have important hyperpolarizing effects (Centonze et al., 2002). By inhibiting the reuptake of the dopamine, bupropion stimulates the condition of the D2 autoreceptors and hyperpolarize the VTA dopaminergic neurons.
A second idea about direct inhibitory effects of bupropion on the VTA dopaminergic neurons, which is considered recently, is the role of dopamine transporter to induce inhibition. This protein with high presynaptic density is a sodium ion- and chloride ion-dependent (Horn, 1990). Its activation by drugs such as amphetamine produces profound hyperpolarization because of inhibiting the sodium influx by cotransport with dopamine. Ionotropic outcomes of dopamine transporter inhibition make it a candidate for the authentic ionotropic-like receptor. The fast in vitro amphetamine-induced currents is comparable with GABAergic ionotropic current. The dopamine transporter in this state couples with chloride shift to enhance the hyperpolarization (DeFelice & Goswami, 2007). The inhibition of the dopamine transporter by increasing the dopamine post-application of bupropion may prevent the influx of sodium that results in hyperpolarization of the VTA-dopaminergic neurons.
The antagonistic effect of the bupropion on nAChRs is the idea based on smoke cessation of the bupropion. The nAChRs are present in the neurons and participate in their excitation and inhibition (Mansvelder, Keath, & McGehee, 2002). The presynaptic nAChRs in the VTA have a functional association with glutamatergic inputs to the VTA and can enhance LTP occurrence in the neurons. Presynaptic excitatory potentials of nAChRs indicate that bupropion can also inhibit the neurons from presynaptic nAChRs antagonism (Mansvelder & McGehee, 2000).
There is evidence of the antagonistic effect of bupropion on α3β2* and α3β4* nAChRs respectively in rats’ striatum and hippocampus across the same concentration range that inhibits dopamine and norepinephrine transporter functions (Miller, Sumithran, & Dwoskin, 2002). The same effect of bupropion can contribute to its inhibitory effect on the neurons. Nicotine or its agonists on nAChRs activation can excite the neurons to release dopamine. nAChR knockout mice express a modified response to nicotine application. The subunits of nAChRs are a key part of the response to nicotine or its antagonist. Activation of β2*-nAChR switches the neurons from a resting state to an excited state (Mameli-Engvall et al., 2006). The study of different nAChRs on neuronal activity reveals that direct governing stimulation and or disinhibition can excite or inhibit the neurons and endogenous acetylcholine in them (Graupner, Maex, & Gutkin, 2013). Bupropion can antagonize and inhibit nAChRs on the neurons with direct and fast inhibition responses.
4.3. Stimulatory effects of bupropion on the VTA dopaminergic neurons: The prominent indirect effect
The stimulatory effects of an antidepressant may arise from the monoamine hypothesis of depression and its pharmacotherapy. The first idea about the mechanism of the stimulatory effects of bupropion on the VTA dopaminergic neurons is based on the overall impact of antidepressants. The stimulatory effects of bupropion may reflect the presence of the stimulatory receptors on the VTA dopaminergic neurons for dopamine because bupropion inhibition of dopamine reuptake can increase dopamine availability to produce excitation. The direct dopamine stimulatory effects on the VTA dopaminergic neurons may be produced by D1-like receptors on the VTA dopaminergic neurons while there is no substantial evidence to support this hypothesis. The density of D1-like receptors on the VTA dopaminergic neurons is very low compared with the density of D2-like receptors.
Direct stimulatory effects of bupropion on the VTA dopaminergic neurons can be due to the action of the dopamine transporter to induce excitation because of its coupling to chloride ions (Horn, 1990). This activity, as seen in the amphetamine fast ionotropic excitatory response to depolarization, is caused by the inhibition of the chloride influx. The fast in vitro amphetamine-induced currents is comparable with glutamatergic ionotropic currents (DeFelice & Goswami, 2007).
The indirect stimulation of the VTA dopaminergic neurons is the most acceptable hypothesis. The GABAergic input to the VTA dopaminergic neurons is one of the major inhibitory inputs, and bupropion alone can reduce the inhibitory GABAergic input to dopaminergic neurons. The VTA dopaminergic neurons are under the tonic discharge of GABAergic inputs from medium spiny neurons of the nucleus accumbens. These neurons exert a tonic inhibitory effect on the VTA dopaminergic neurons and suppress their activity (Haber, Wolfe, & Groenewegen, 1990; Kalivas, Churchill, & Klitenick, 1993; Nauta, Smith, Faull, & Domesick, 1978). The nucleus accumbens has a noticeable effect on the neuronal activity of VTA and VTA-nucleus accumbens interaction has a key role in the firing rate (Moaddab, Kermani, Azizi, & Haghparast, 2013).
On the other hand, bupropion as a nicotinic acetylcholine receptors (nAChRs) antagonist can inhibit nicotine effects on GABA neurons, and the nAChRs blockade can remove the excitatory drive or disinhibition to GABA neurons (Fryer & Lukas, 1999; Slemmer, Martin, & Damaj, 2000). The GABAergic innervation blockade enhances neuronal activity with a disinhibition mechanism (Johnson & North, 1992). The nAChRs in the VTA neurons can also contribute to their nicotine rewarding effects (Nisell, Nomikos, & Svensson, 1994).
The excitation of VTA dopaminergic neurons by acute micro-infusion of bupropion is consistent with the emerging views on the dopamine role in reward-based learning and memory processes (Berridge, 2007; De Biasi & Dani, 2011; Schultz, 2007). Increase in neuronal activity of VTA can amplify and synchronize the output of DA signaling to different targets such as the nucleus accumbens. Recent studies show that VTA neuronal activity enhances rewards and correlates with the increase in synchronization of neuronal spiking (Cryan, Bruijnzeel, Skjei, & Markou, 2003; De Biasi & Dani, 2011; Joshua et al., 2009).
Inhibition of the dopamine reuptake can increase the synaptic dopamine content. The onset of bupropion excitatory effect on the neurons has many delays in comparison with its inhibitory effects. This difference indicates that excitation could be generated by afferent neurons, not by their somatodendritic receptors. Increase in discharge rate by bupropion can also be explained by a decrease in tonic inhibitory inputs to the VTA. In this study, the changes in neuronal activity, excitation, and inhibition began 60±10.5 s and 8±4.4 s respectively after the maximum amount of bupropion. This delay in the onset of bupropion effects suggests that its excitatory effects on the neurons are carried out via indirect effect or probable disinhibition of GABAergic inputs to the VTA. We propose that different time courses from inactivation of nAChR subtypes on GABA inputs to the neurons can contribute to the bupropion-delayed excitatory effect. The distinct contributions of different nAChRs in VTA dopaminergic neurons have been already defined in many studies. In addition to nicotine, activation of α4α6β2* nAChRs in the neurons is sufficient to support the initiation of cellular changes. α4α6β2* nAChRs may be a promising target for future drug addiction (Engle, Shih, McIntosh, & Drenan, 2013). The postulated differences in distinct contributions of α4* and α6* nAChRs in the soma vs. the axon terminal of nucleus accumbens neurons are related to neuronal firing properties (Exley et al., 2011).
4.4. Inhibitory effects of bupropion on the VTA dopaminergic neurons: The prominent direct effect
Based on our study results, the inhibitory effects of the microiontophoretic application of bupropion were much stronger than its excitatory effects on VTA dopaminergic neuronal activity. Several possible mechanisms can cause inhibitory effects.
The direct inhibitory effects of the bupropion on VTA dopaminergic neurons are due to the occupation of the D2-like dopamine receptors. The major D2-like dopamine receptors are autoreceptors on the somatodendritic regions and strongly inhibit the VTA-dopaminergic neurons by lowering intracellular cAMP. Lejeune and Millan (1995) reported that the systemic application of the D3 dopamine receptor selective agonists could inhibit VTA dopaminergic neuronal firing rate that is blocked by D3 dopamine receptor antagonists’ dose-dependently both of them (Lejeune & Millan, 1995). The activation of the D2 dopamine receptors on the VTA dopaminergic neurons in the wild-type mice could produce profound hyperpolarization, but in the D2-/- mice, it was abolished due to receptor lacking. However, the subtype of the D2 dopamine receptor is essential because of its response to activation. On the other hand, the overall results show that the D2 dopamine autoreceptors on the VTA dopaminergic neurons have important hyperpolarizing effects (Centonze et al., 2002). By inhibiting the reuptake of the dopamine, bupropion stimulates the condition of the D2 autoreceptors and hyperpolarize the VTA dopaminergic neurons.
A second idea about direct inhibitory effects of bupropion on the VTA dopaminergic neurons, which is considered recently, is the role of dopamine transporter to induce inhibition. This protein with high presynaptic density is a sodium ion- and chloride ion-dependent (Horn, 1990). Its activation by drugs such as amphetamine produces profound hyperpolarization because of inhibiting the sodium influx by cotransport with dopamine. Ionotropic outcomes of dopamine transporter inhibition make it a candidate for the authentic ionotropic-like receptor. The fast in vitro amphetamine-induced currents is comparable with GABAergic ionotropic current. The dopamine transporter in this state couples with chloride shift to enhance the hyperpolarization (DeFelice & Goswami, 2007). The inhibition of the dopamine transporter by increasing the dopamine post-application of bupropion may prevent the influx of sodium that results in hyperpolarization of the VTA-dopaminergic neurons.
The antagonistic effect of the bupropion on nAChRs is the idea based on smoke cessation of the bupropion. The nAChRs are present in the neurons and participate in their excitation and inhibition (Mansvelder, Keath, & McGehee, 2002). The presynaptic nAChRs in the VTA have a functional association with glutamatergic inputs to the VTA and can enhance LTP occurrence in the neurons. Presynaptic excitatory potentials of nAChRs indicate that bupropion can also inhibit the neurons from presynaptic nAChRs antagonism (Mansvelder & McGehee, 2000).
There is evidence of the antagonistic effect of bupropion on α3β2* and α3β4* nAChRs respectively in rats’ striatum and hippocampus across the same concentration range that inhibits dopamine and norepinephrine transporter functions (Miller, Sumithran, & Dwoskin, 2002). The same effect of bupropion can contribute to its inhibitory effect on the neurons. Nicotine or its agonists on nAChRs activation can excite the neurons to release dopamine. nAChR knockout mice express a modified response to nicotine application. The subunits of nAChRs are a key part of the response to nicotine or its antagonist. Activation of β2*-nAChR switches the neurons from a resting state to an excited state (Mameli-Engvall et al., 2006). The study of different nAChRs on neuronal activity reveals that direct governing stimulation and or disinhibition can excite or inhibit the neurons and endogenous acetylcholine in them (Graupner, Maex, & Gutkin, 2013). Bupropion can antagonize and inhibit nAChRs on the neurons with direct and fast inhibition responses.
The precise response of the neurons to the route of the drug application is vital for its design and time of the delivery. Researchers have recently focused on the pharmacological responses of the agents on the reward system because of their wide spectrum use and abuse in the world. The VTA dopaminergic neurons that can produce a massive and functional dopaminergic outflow of the brain are the core part of this focus. However, in contrast to some dopaminergic nuclei, about 55% of neurons in the VTA are dopaminergic and putatively selected neurons can reveal the heterogeneity of VTA neuronal response. The high density of D2 dopamine receptors on the somatodendritic region of the VTA dopaminergic neurons has a critical functional modulatory effect on these neurons. Evidence of inhibitory and excitatory effects of bupropion with its direct local application reveal that temporal and spatial availability is essential for its nature of effect.
In summary, this study presents new data on bupropion effects on the VTA dopaminergic neuronal firing rate, whereas many previous studies have administered bupropion systemically and only explained its general action. Our obtained results emphasize that bupropion can excite or inhibit the neurons, while its direct effect on them is inhibitory.
Ethical Considerations
Compliance with ethical guidelines
Urmia Medical Science Research Ethics Committee (UMSREC) reviewed all procedures and experiments as the local referral Biomedical Committee for Research Ethics. Protocols and guidelines were carried on in accordance with the National Institutes of Health (NIH) for the care and use of experimental animals. The Animal Laboratory Center of Urmia University of Medical Sciences precisely outlined these protocols and guidelines. Supervision of animal protocols was carried out according to the research design.
Funding
Urmia Research and Technology Vice Chancellor Grant (Grant No. 1647 and 1962) sponsored this study and the practical works were done at Danesh Pey Hadi Company lab.
Authors' contributions
Study concept, design, student mentorship: Firouz Ghaderi Pakdel; Acquisition of animal data: Shirin Sadighparvar and Fereshteh Tale; Igor analysis blindly: Somayyeh Naderi; Data analysis, interpretation of the findings: Parviz Shahabi; and Reviewing the manuscript and approving the final version for publication: All authors.
Conflict of interest
The authors declare no conflict of interest regarding this study and manuscript preparation after reading the Journal’s position on issues concerning the ethical publication and confirm that this report is consistent with those guidelines.
Acknowledgments
The study procedure was performed in Danesh Pey Hadi Company as knowledge base Company of Health Technology Development Center at Urmia University of Medical Sciences.
References
In summary, this study presents new data on bupropion effects on the VTA dopaminergic neuronal firing rate, whereas many previous studies have administered bupropion systemically and only explained its general action. Our obtained results emphasize that bupropion can excite or inhibit the neurons, while its direct effect on them is inhibitory.
Ethical Considerations
Compliance with ethical guidelines
Urmia Medical Science Research Ethics Committee (UMSREC) reviewed all procedures and experiments as the local referral Biomedical Committee for Research Ethics. Protocols and guidelines were carried on in accordance with the National Institutes of Health (NIH) for the care and use of experimental animals. The Animal Laboratory Center of Urmia University of Medical Sciences precisely outlined these protocols and guidelines. Supervision of animal protocols was carried out according to the research design.
Funding
Urmia Research and Technology Vice Chancellor Grant (Grant No. 1647 and 1962) sponsored this study and the practical works were done at Danesh Pey Hadi Company lab.
Authors' contributions
Study concept, design, student mentorship: Firouz Ghaderi Pakdel; Acquisition of animal data: Shirin Sadighparvar and Fereshteh Tale; Igor analysis blindly: Somayyeh Naderi; Data analysis, interpretation of the findings: Parviz Shahabi; and Reviewing the manuscript and approving the final version for publication: All authors.
Conflict of interest
The authors declare no conflict of interest regarding this study and manuscript preparation after reading the Journal’s position on issues concerning the ethical publication and confirm that this report is consistent with those guidelines.
Acknowledgments
The study procedure was performed in Danesh Pey Hadi Company as knowledge base Company of Health Technology Development Center at Urmia University of Medical Sciences.
References
- Albanese, A., & Minciacchi, D. (1983). Organization of the ascending projections from the ventral tegmental area: A multiple fluorescent retrograde tracer study in the rat. Journal of Comparative Neurology, 216(4), 406-20. [DOI:10.1002/cne.902160406] [PMID]
- Amirabadi, S., Pakdel, F. G., Shahabi, P., Naderi, S., Osalou, M. A., & Cankurt, U. (2014). Microinfusion of bupropion inhibits putative GABAergic neuronal activity of the ventral tegmental area. Basic and Clinical Neuroscience, 5(3), 182-90. [PMID] [PMCID]
- Ascher, J. A., Cole, J. O., Colin, J. N., Feighner, J. P., Ferris, R. M., Fibiger, H. C., et al. (1995). Bupropion: A review of its mechanism of antidepressant activity. The Journal of Clinical Psychiatry, 56(9), 395-401. [PMID]
- Beaulieu, J. M., & Gainetdinov, R. R. (2011). The physiology, signaling, and pharmacology of dopamine receptors. Pharmacological Reviews, 63(1), 182-217. [DOI:10.1124/pr.110.002642] [PMID]
- Berridge, K. C. (2007). The debate over dopamine’s role in reward: The case for incentive salience. Psychopharmacology, 191(3), 391-431. [DOI:10.1007/s00213-006-0578-x] [PMID]
- Besson, M., David, V., Baudonnat, M., Cazala, P., Guilloux, J. P., Reperant, C., et al. (2012). Alpha7-nicotinic receptors modulate nicotine-induced reinforcement and extracellular dopamine outflow in the mesolimbic system in mice. Psychopharmacology, 220(1), 1-14. [DOI:10.1007/s00213-011-2422-1] [PMID]
- Brown, A. S., & Gershon, S. (1993). Dopamine and depression. Journal of Neural Transmission, 91(2-3), 75-109. [DOI:10.1007/BF01245227]
- Cameron, D. L., Wessendorf, M. W., & Williams, J. T. (1997). A subset of ventral tegmental area neurons is inhibited by dopamine, 5-hydroxytryptamine and opioids. Neuroscience, 77(1), 155-66. [DOI:10.1016/S0306-4522(96)00444-7]
- Centonze, D., Usiello, A., Gubellini, P., Pisani, A., Borrelli, E., Bernardi, G., et al. (2002). Dopamine D2 receptor-mediated inhibition of dopaminergic neurons in mice lacking D2L receptors. Neuropsychopharmacology, 27(5), 723-6. [DOI:10.1016/S0893-133X(02)00367-6]
- Cooper, B. R., Wang, C. M., Cox, R. F., Norton, R., Shea, V., & Ferris, R. M. (1994). Evidence that the acute behavioral and electrophysiological effects of bupropion (Wellbutrin) are mediated by a noradrenergic mechanism. Neuropsychopharmacology, 11(2), 133-41. [DOI:10.1038/npp.1994.43] [PMID]
- Cryan, J. F., Bruijnzeel, A. W., Skjei, K. L., & Markou, A. (2003). Bupropion enhances brain reward function and reverses the affective and somatic aspects of nicotine withdrawal in the rat. Psychopharmacology, 168(3), 347-58. [DOI:10.1007/s00213-003-1445-7] [PMID]
- De Biasi, M., & Dani, J. A. (2011). Reward, addiction, withdrawal to nicotine. Annual Review of Neuroscience, 34, 105-30. [DOI:10.1146/annurev-neuro-061010-113734] [PMID] [PMCID]
- DeFelice, L. J., & Goswami, T. (2007). Transporters as channels. Annual Review of Physiology, 69, 87-112. [DOI:10.1146/annurev.physiol.69.031905.164816] [PMID]
- Diehl, D. J., & Gershon, S. (1992). The role of dopamine in mood disorders. Comprehensive Psychiatry, 33(2), 115-120. [DOI:10.1016/0010-440X(92)90007-D]
- Dong, J., & Blier, P. (2001). Modification of norepinephrine and serotonin, but not dopamine, neuron firing by sustained bupropion treatment. Psychopharmacology, 155(1), 52-57. [DOI:10.1007/s002130000665]
- Dremencov, E., Gispan-Herman, I., Rosenstein, M., Mendelman, A., Overstreet, D. H., Zohar, J., et al. (2004). The serotonin-dopamine interaction is critical for fast-onset action of antidepressant treatment: In vivo studies in an animal model of depression. Psychopharmacology & Biological Psychiatry, 28(1), 141-7. [DOI:10.1016/j.pnpbp.2003.09.030] [PMID]
- Dremencov, E., Newman, M. E., Kinor, N., Blatman-Jan, G., Schindler, C. J., Overstreet, D. H., et al. (2005). Hyperfunctionality of serotonin-2C receptor-mediated inhibition of accumbal dopamine release in an animal model of depression is reversed by antidepressant treatment. Neuropharmacology, 48(1), 34-42. [DOI:10.1016/j.neuropharm.2004.09.013] [PMID]
- El Mansari, M., Ghanbari, R., Janssen, S., & Blier, P. (2008). Sustained administration of bupropion alters the neuronal activity of serotonin, norepinephrine but not dopamine neurons in the rat brain. Neuropharmacology, 55(7), 1191-8. [DOI:10.1016/j.neuropharm.2008.07.028] [PMID]
- Engle, S. E., Shih, P. Y., McIntosh, J. M., & Drenan, R. M. (2013). α4α6β2* nicotinic acetylcholine receptor activation on ventral tegmental area dopamine neurons is sufficient to stimulate a depolarizing conductance and enhance surface AMPA receptor function. Molecular Pharmacology, 84(3), 393-406. [DOI:10.1124/mol.113.087346] [PMID] [PMCID]
- Exley, R., Maubourguet, N., David, V., Eddine, R., Evrard, A., Pons, S., et al. (2011). Distinct contributions of nicotinic acetylcholine receptor subunit α4 and subunit α6 to the reinforcing effects of nicotine. Proceedings of the National Academy of Sciences, 108(18), 7577-82. [DOI:10.1073/pnas.1103000108] [PMID] [PMCID]
- Fabre, L. F., Brodie, H. K., Garver, D., & Zung, W. W. (1983). A multicenter evaluation of bupropion versus placebo in hospitalized depressed patients. The Journal of Clinical Psychiatry, 44(5 Pt 2), 88-94. [PMID]
- Fallon, J. H. (1988). Topographic organization of ascending dopaminergic projections. Annals of the New York Academy of Sciences, 537(1), 1-9. [DOI:10.1111/j.1749-6632.1988.tb42093.x] [PMID]
- Fryer, J. D., & Lukas, R. J. (1999). Noncompetitive functional inhibition at diverse, human nicotinic acetylcholine receptor subtypes by bupropion, phencyclidine, and ibogaine. Journal of Pharmacology and Experimental Therapeutics, 288(1), 88-92. [PMID]
- Ghanbari, R., El Mansari, M., & Blier, P. (2010). Electrophysiological effects of the co-administration of escitalopram and bupropion on rat serotonin and norepinephrine neurons. Journal of Psychopharmacology, 24(1), 39-50. [DOI:10.1177/0269881108095714] [PMID]
- Graupner, M., Maex, R., & Gutkin, B. (2013). Endogenous cholinergic inputs and local circuit mechanisms govern the phasic mesolimbic dopamine response to nicotine. PLOS Computational Biology, 9(8), e1003183. [DOI:10.1371/journal.pcbi.1003183] [PMID] [PMCID]
- Green, T. R. (1997). Bupropion for SSRI-induced fatigue. The Journal of Clinical Psychiatry, 58(4), 174. [DOI:10.4088/JCP.v58n0407a] [PMID]
- Haber, S. N., Wolfe, D. P., & Groenewegen, H. J. (1990). The relationship between ventral striatal efferent fibers and the distribution of peptide-positive woolly fibers in the forebrain of the rhesus monkey. Neuroscience, 39(2), 323-38. [DOI:10.1016/0306-4522(90)90271-5]
- Hnasko, T. S., Hjelmstad, G. O., Fields, H. L., & Edwards, R. H. (2012). Ventral tegmental area glutamate neurons: Electrophysiological properties and projections. Journal of Neuroscience, 32(43), 15076-85. [DOI:10.1523/JNEUROSCI.3128-12.2012] [PMID] [PMCID]
- Holly, E. N., & Miczek, K. A. (2016). Ventral tegmental area dopamine revisited: Effects of acute and repeated stress. Psychopharmacology, 233(2), 163-86. [DOI:10.1007/s00213-015-4151-3] [PMID] [PMCID]
- Horn, A. S. (1990). Dopamine uptake: A review of progress in the last decade. Progress in Neurobiology, 34(5), 387-400. [DOI:10.1016/0301-0082(90)90033-D]
- Horst, W. D., & Preskorn, S. H. (1998). Mechanisms of action and clinical characteristics of three atypical antidepressants: Venlafaxine, nefazodone, bupropion. Journal of Affective Disorders, 51(3), 237-54. [DOI:10.1016/S0165-0327(98)00222-5]
- Jahanbani, M., Nasri, S., Pakdel, F. G., Cankurt, U., Shahabi, P., Amirabadi, S., et al. (2014). The effect of acute intra Locus Coeruleus (LC) microinfusion of bupropion on formalin-induced pain behavior in rat. Basic and Clinical Neuroscience, 5(1), 31-41. [PMID] [PMCID]
- Johnson, S. W., & North, R. A. (1992). Two types of neurone in the rat ventral tegmental area and their synaptic inputs. The Journal of Physiology, 450(1), 455-68. [DOI:10.1113/jphysiol.1992.sp019136] [PMID] [PMCID]
- Joshua, M., Adler, A., Prut, Y., Vaadia, E., Wickens, J. R., & Bergman, H. (2009). Synchronization of midbrain dopaminergic neurons is enhanced by rewarding events. Neuron, 62(5), 695-704. [DOI:10.1016/j.neuron.2009.04.026] [PMID]
- Kalivas, P. W. (1993). Neurotransmitter regulation of dopamine neurons in the ventral tegmental area. Brain Research Reviews, 18(1), 75-113. [DOI:10.1016/0165-0173(93)90008-N]
- Kalivas, P. W., Churchill, L., & Klitenick, M. A. (1993). GABA and enkephalin projection from the nucleus accumbens and ventral pallidum to the ventral tegmental area. Neuroscience, 57(4), 1047-60. [DOI:10.1016/0306-4522(93)90048-K]
- Klink, R., de Kerchove d’Exaerde, A., Zoli, M., & Changeux, J. P. (2001). Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. Journal of Neuroscience, 21(5), 1452-63. [DOI:10.1523/JNEUROSCI.21-05-01452.2001] [PMID]
- Lammel, S., Hetzel, A., Hackel, O., Jones, I., Liss, B., & Roeper, J. (2008). Unique properties of mesoprefrontal neurons within a dual mesocorticolimbic dopamine system. Neuron, 57(5), 760-773. [DOI:10.1016/j.neuron.2008.01.022] [PMID]
- Le Moal, M., & Simon, H. (1991). Mesocorticolimbic dopaminergic network: functional and regulatory roles. Physiological Reviews, 71(1), 155-234. [DOI:10.1152/physrev.1991.71.1.155] [PMID]
- Lejeune, F., & Millan, M. J. (1995). Activation of dopamine D3 autoreceptors inhibits firing of ventral tegmental dopaminergic neurones in vivo. European Journal of Pharmacology, 275(3), R7-R9. [DOI:10.1016/0014-2999(95)00106-U]
- Mameli-Engvall, M., Evrard, A., Pons, S., Maskos, U., Svensson, T. H., Changeux, J. P., et al. (2006). Hierarchical control of dopamine neuron-firing patterns by nicotinic receptors. Neuron, 50(6), 911-21. [DOI:10.1016/j.neuron.2006.05.007] [PMID]
- Mansvelder, H. D., & McGehee, D. S. (2000). Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron, 27(2), 349-57. [DOI:10.1016/S0896-6273(00)00042-8]
- Mansvelder, H. D., Fagen, Z. M., Chang, B., Mitchum, R., & McGehee, D. S. (2007). Bupropion inhibits the cellular effects of nicotine in the ventral tegmental area. Biochemical Pharmacology, 74(8), 1283-91. [DOI:10.1016/j.bcp.2007.07.034] [PMID] [PMCID]
- Mansvelder, H. D., Keath, J. R., & McGehee, D. S. (2002). Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron, 33(6), 905-19. [DOI:10.1016/S0896-6273(02)00625-6]
- Marinelli, M., Cooper, D. C., Baker, L. K., & White, F. J. (2003). Impulse activity of midbrain dopamine neurons modulates drug-seeking behavior. Psychopharmacology, 168(1-2), 84-98. [DOI:10.1007/s00213-003-1491-1] [PMID]
- Mejías-Aponte, C. A., & Kiyatkin, E. A. (2012). Ventral tegmental area neurons are either excited or inhibited by cocaine’s actions in the peripheral nervous system. Neuroscience, 207, 182-97. [DOI:10.1016/j.neuroscience.2012.01.026] [PMID] [PMCID]
- Miller, D. K., Sumithran, S. P., & Dwoskin, L. P. (2002). Bupropion inhibits nicotine-evoked [3H] overflow from rat striatal slices preloaded with [3H] dopamine and from rat hippocampal slices preloaded with [3H] norepinephrine. Journal of Pharmacology and Experimental Therapeutics, 302(3), 1113-22. [DOI:10.1124/jpet.102.033852] [PMID]
- Moaddab, M., Kermani, M., Azizi, P., & Haghparast, A. (2013). Functional interaction between the shell sub-region of the nucleus accumbens and the ventral tegmental area in response to morphine: An electrophysiological study. Basic and Clinical Neuroscience, 4(2), 159-60. [PMID] [PMCID]
- Moritz, A. E., Free, R. B., & Sibley, D. R. (2018). Advances and challenges in the search for D2 and D3 dopamine receptor-selective compounds. Cellular Signalling, 41, 75-81. [DOI:10.1016/j.cellsig.2017.07.003] [PMID] [PMCID]
- Naderi, S., Pakdel, F. G., Osalou, M. A., & Cankurt, U. (2014). Acute systemic infusion of bupropion decrease formalin induced pain behavior in rat. The Korean Journal of Pain, 27(2), 118-24. [DOI:10.3344/kjp.2014.27.2.118] [PMID] [PMCID]
- Nair-Roberts, R. G., Chatelain-Badie, S. D., Benson, E., White-Cooper, H., Bolam, J. P., & Ungless, M. A. (2008). Stereological estimates of dopaminergic, GABAergic and glutamatergic neurons in the ventral tegmental area, substantia nigra and retrorubral field in the rat. Neuroscience, 152(4), 1024-31. [DOI:10.1016/j.neuroscience.2008.01.046] [PMID] [PMCID]
- Nauta, W. J. H., Smith, G. P., Faull, R. L. M., & Domesick, V. B. (1978). Efferent connections and nigral afferents of the nucleus accumbens septi in the rat. Neuroscience, 3(4-5), 385-401. [DOI:10.1016/0306-4522(78)90041-6]
- Nisell, M., Nomikos, G. G., & Svensson, T. H. (1994). Systemic nicotine‐induced dopamine release in the rat nucleus accumbens is regulated by nicotinic receptors in the ventral tegmental area. Synapse, 16(1), 36-44. [DOI:10.1002/syn.890160105] [PMID]
- Nutt, D. J., Baldwin, D. S., Clayton, A. H., Elgie, R., Lecrubier, Y., Montejo, A. L., et al. (2006). Consensus statement and research needs: The role of dopamine and norepinephrine in depression and antidepressant treatment. The Journal of Clinical Psychiatry, 67(Suppl. 6), 46-49. [PMID]
- Pakdel, F. G., Amirabadi, S., Naderi, S., Osalou, M. A., Cankurt, U., Jahanbani, M., et al. (2014). Effects of acute intracerebroventricular microinfusions of bupropion on background spike activity of locus coeruleus neurons in rats. Neurophysiology, 46(4), 316-22. [DOI:10.1007/s11062-014-9450-5]
- Paxinos, G., & Watson, C. (2007). The rat brain in stereotaxic coordinates. 6th Ed. Cambridge: Academic Press.
- Pereira, V. M., Arias-Carrion, O., Machado, S., Nardi, A. E., & Silva, A. C. (2014). Bupropion in the depression-related sexual dysfunction: A systematic review. CNS & Neurological Disorders-Drug Targets, 13(6), 1079-88. [DOI:10.2174/1871527313666140612112630] [PMID]
- Randrup, A., & Braestrup, C. (1977). Uptake inhibition of biogenic amines by newer antidepressant drugs: Relevance to the dopamine hypothesis of depression. Psychopharmacology, 53(3), 309-14. [DOI:10.1007/BF00492370]
- Sampson, D., Willner, P., & Muscat, R. (1991). Reversal of antidepressant action by dopamine antagonists in an animal model of depression. Psychopharmacology, 104(4), 491-95. [DOI:10.1007/BF02245655]
- Schildkraut, J. J. (1967). The catecholamine hypothesis of affective disorders: A review of supporting evidence. American Journal of Psychiatry, 4(3), 203-17. [DOI:10.1176/AJP.122.5.509]
- Schultz, W. (2007). Multiple dopamine functions at different time courses. Annual Review of Neuroscience, 30, 259-88. [DOI:10.1146/annurev.neuro.28.061604.135722] [PMID]
- Sidhpura, N., Redfern, P., Rowley, H., Heal, D., & Wonnacott, S. (2007). Comparison of the effects of bupropion and nicotine on locomotor activation and dopamine release in vivo. Biochemical Pharmacology, 74(8), 1292-8. [DOI:10.1016/j.bcp.2007.06.025] [PMID]
- Slemmer, J. E., Martin, B. R., & Damaj, M. I. (2000). Bupropion is a nicotinic antagonist. Journal of Pharmacology and Experimental Therapeutics, 295(1), 321-7. [PMID]
- Spanagel, R., Herz, A., & Shippenberg, T. S. (1992). Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proceedings of the National Academy of Sciences, 89(6), 2046-50. [DOI:10.1073/pnas.89.6.2046] [PMID] [PMCID]
- Spano, P., Govoni, S., & Trabucchi, M. (1978). Studies on the pharmacological properties of dopamine receptors in various areas of the central nervous system. Advances in Biochemical Psychopharmacology, 19, 155-65. [PMID]
- Stuber, G. D., Hnasko, T. S., Britt, J. P., Edwards, R. H., & Bonci, A. (2010). Dopaminergic terminals in the nucleus accumbens but not the dorsal striatum corelease glutamate. Journal of Neuroscience, 30(24), 8229-33. [DOI:10.1523/JNEUROSCI.1754-10.2010] [PMID] [PMCID]
- Taeron, C. (2002). [Bupropion. An antidepressant used to help smoking cessation (French)]. Revue de l'infirmiere, (81), 47-49.
- Ungless, M. A., & Grace, A. A. (2012). Are you or aren’t you? Challenges associated with physiologically identifying dopamine neurons. Trends in Neurosciences, 35(7), 422-30. [DOI:10.1016/j.tins.2012.02.003] [PMID] [PMCID]
- West, C. H., & Weiss, J. M. (2011). Effects of chronic antidepressant drug administration and electroconvulsive shock on activity of dopaminergic neurons in the ventral tegmentum. The International Journal of Neuropsychopharmacology, 14(2), 201-10. [DOI:10.1017/S1461145710000489] [PMID] [PMCID]
- White, F. J., & Wang, R. Y. (1984). Electrophysiological evidence for A10 dopamine autoreceptor subsensitivity following chronicd-amphetamine treatment. Brain Research, 309(2), 277-82. [DOI:10.1016/0006-8993(84)90594-8]
- Willner, P. (1983). Dopamine and depression: A review of recent evidence. III. The effects of antidepressant treatments. Brain Research Reviews, 6(3), 237-46. [DOI:10.1016/0165-0173(83)90007-3]
- Wooltorton, J. R., Pidoplichko, V. I., Broide, R. S., & Dani, J. A. (2003). Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. Journal of Neuroscience, 23(8), 3176-85. [DOI:10.1523/JNEUROSCI.23-08-03176.2003] [PMID]
- Yamaguchi, T., Wang, H. L., Li, X., Ng, T. H., & Morales, M. (2011). Mesocorticolimbic glutamatergic pathway. Journal of Neuroscience, 31(23), 8476-90. [DOI:10.1523/JNEUROSCI.1598-11.2011] [PMID]
- Zhang, T. A., Placzek, A. N., & Dani, J. A. (2010). In vitro identification and electrophysiological characterization of dopamine neurons in the ventral tegmental area. Neuropharmacology, 59(6), 431-6. [DOI:10.1016/j.neuropharm.2010.06.004] [PMID] [PMCID]
Type of Study: Original |
Subject:
Behavioral Neuroscience
Received: 2017/10/10 | Accepted: 2018/04/21 | Published: 2019/07/1
Received: 2017/10/10 | Accepted: 2018/04/21 | Published: 2019/07/1
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
